De novo-designed light-activated Reaction Centers pave the way toward new photosynthetic pathways
Published in Bioengineering & Biotechnology
Photosynthesis provides the chemical energy that powers the global food web. When it comes to human energy needs, however, photosynthetic biofuel production is inefficient compared to other renewable energy technologies. Photosynthetic organisms face a ~12% theoretical limit for solar-to-glucose conversion efficiency due to the two-photosystem Z-scheme of oxygenic photosynthesis in which Photosystems I and II compete against each other for photons in the same wavelength ranges1. In the fight against climate change and for food and energy security, we will need to abandon the two-photosystem model and redesign the overall architecture of photosynthesis before we can achieve high efficiency solar-to-fuel energy conversion in living cells that beats the 12% efficiency limit.
Electron transfer theory tells us that in principle, natural reaction center (RC) proteins can be greatly improved. An RC is a protein that initiates the light-activated electron transfer cascade of photosynthesis. It uses electron donors (D), a pigment (P), and electron acceptors (A) to make a DPA electron transport chain that separates charge, as illustrated in Figure 1.
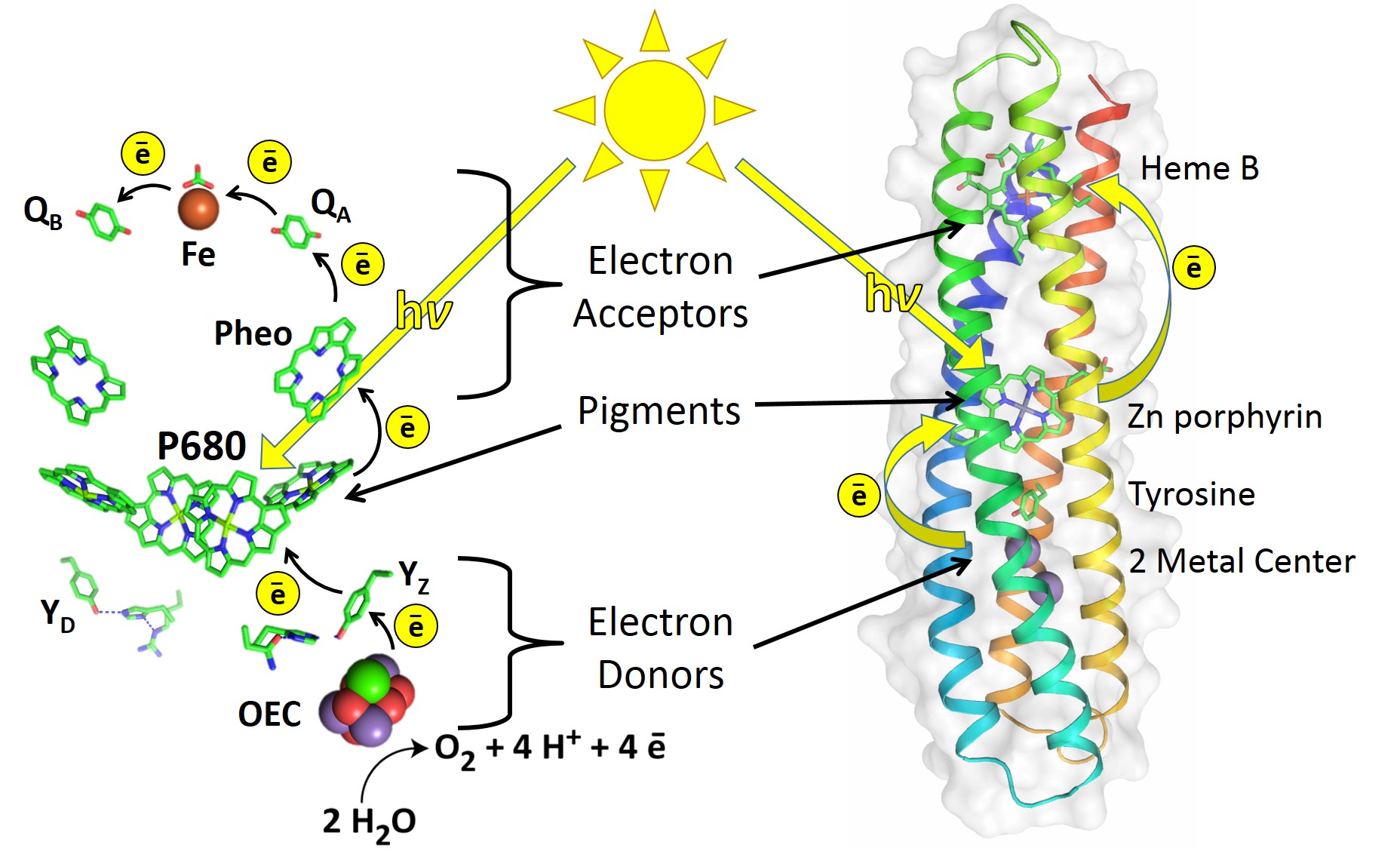
Figure 1: The RC of Photosystem II (left) uses electron donors, pigments, and electron acceptors for a series of electron transfer reactions2. Similarly, the de novo-designed RC maquette protein (right) assembles a DPA electron transport chain that uses light energy to drive photochemical charge separation.
To create higher efficiency RCs, we would need to radically reengineer native photosystems or, better yet, design completely new RCs from scratch. In this paper, we take a first step toward building a new photosynthetic pathway from first principles. We describe the design, X-ray crystal structures, and electron transfer activity of an RC maquette protein. “Maquettes” are small, robust, de novo-designed proteins meant to simplify the challenge of elucidating structure-function relationships in proteins. Protein maquettes let us put our theories to the test; they play a similar role to scale maquettes that architects use to evaluate their designs.
We designed the RC maquette to assemble redox centers with inter-cofactor distances appropriate for multi-step charge separation. By applying a well-tested empirical equation to predict electron transfer rates3, we selected donor, pigment, and acceptor positions in the RC maquette that would facilitate charge separation for a wide range of electron transfer free energies.
Once we identified the usable inter-cofactor distances for our electron transport chain, we looked to natural proteins that could be used as guides in the design of the RC maquette. The cytochrome b subunits of cytochromes bc1 and b6f are examples of porphyrin-binding proteins in which the inter-cofactor spacing would be appropriate for electron tunneling in the RC maquette. Bacterioferritin and other diiron proteins (including some de novo-designed examples4,5) model the assembly of tyrosine and metal ions as electron donors. Photosystem II demonstrates the use of a histidine-tyrosine hydrogen bond in an electron transport chain. Without copying amino acid sequences from nature, we designed a four-helix bundle maquette that recreates structural elements of all these proteins to assemble a working RC. Crystal structures show that the RC maquette successfully reproduces the intended structural motifs. Figure 2 compares the electron acceptor site of the RC maquette to cytochrome b.
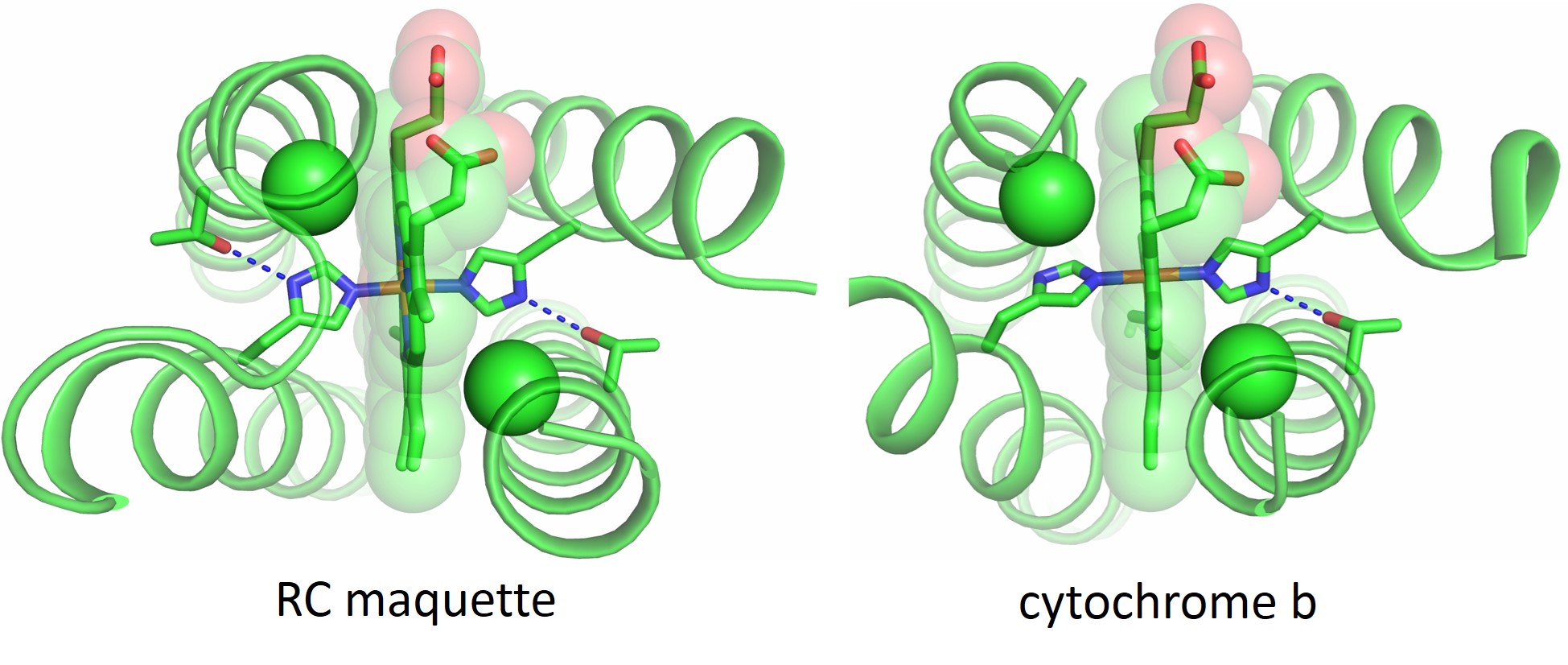
Figure 2: Crystal structures show that the electron acceptor module of the RC maquette (left, PDB ID: 5VJT) resembles cytochrome bc1 (right, PDB ID: 2A06)6 despite low sequence identity between the structures. Notch glycine Cα atoms are shown as green spheres and Thr-His hydrogen bonds are shown as blue dotted lines.
In order for an RC to be useful, it must form a D+PA- charge separated state that persists long enough to be used for productive chemistry. One-electron transfer steps of water oxidation in Photosystem II take as long as 1-2 milliseconds7, but the longest charge separation lifetimes in synthetic chemical DPA triads only last for tens of microseconds8.
To determine whether the RC maquette can form long-lasting charge separated states, we used transient absorption spectroscopy, which lets us monitor time-resolved changes in color that are indicative of the electronic state of the system. When a Zn porphyrin pigment binds to the RC maquette in the absence of electron donors or acceptors, transient absorption spectroscopy detects a long-lived pigment excited state following light absorption. Addition of an electron-accepting iron- or cobalt-porphyrin to make a PA dyad shortens the pigment excited state lifetime 50-fold due to electron transfer, but charge recombination is too fast in the dyads to allow the charge separated state to be useful for chemical catalysis. However, the inclusion of an electron donor to make a complete DPA triad enables long-lived charge separation; the pigment photoreduces the acceptor, and the donor reduces the oxidized pigment before charge recombination can take place. In the tyrosine-Zn porphyrin-heme triad shown in Figure 3, we measured a charge separated state lasting for 3 ms with a quantum yield of 11%.
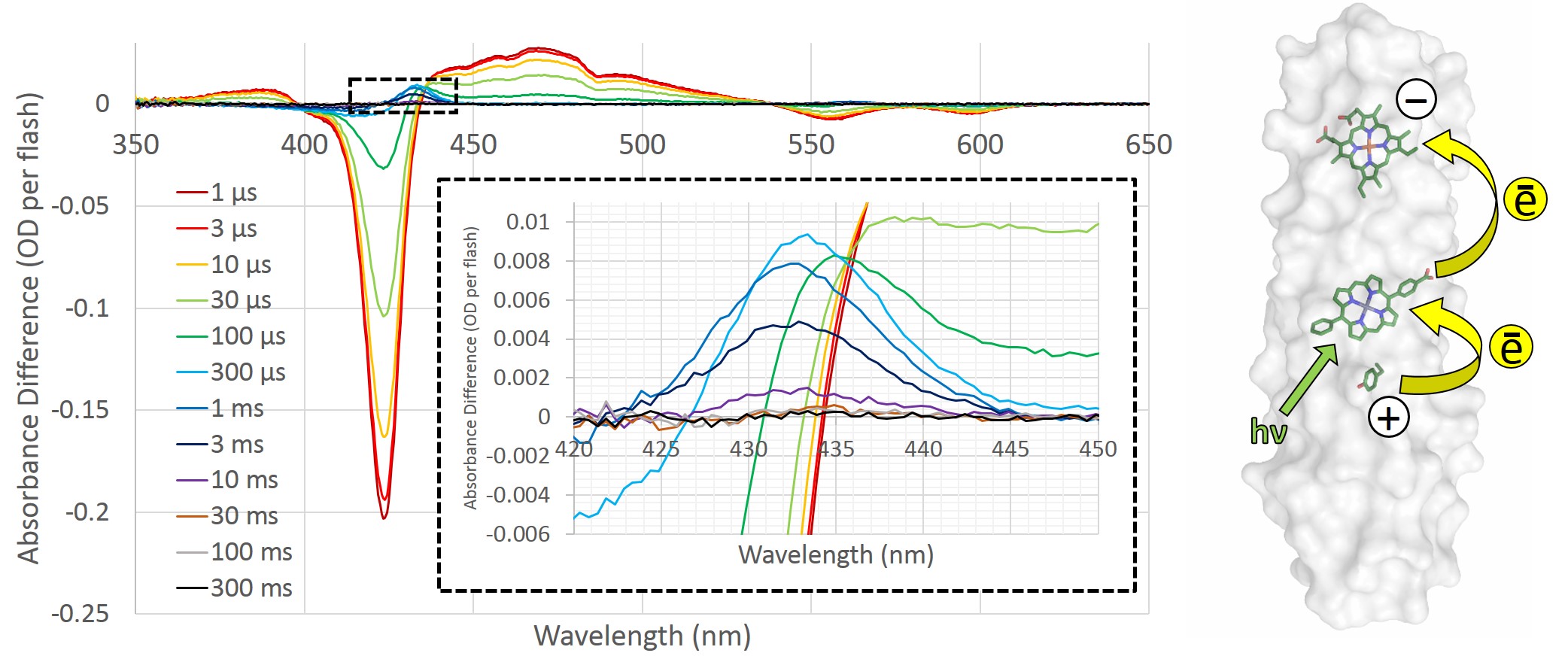
Figure 3: Transient absorption spectroscopy of the tyrosine-Zn porphyrin-heme triad. The intense Zn porphyrin Soret bleach at 422 nm indicates photoexcitation of the pigment. Inset: A close-up of the heme redox signal at 432 nm, which enables estimation of the yield and lifetime of charge separation. The presence of tyrosine168 makes the heme redox signal at 432 nm detectable for milliseconds; removal of the electron donor by mutation of tyrosine to leucine eliminates the 432 nm signal. Right: a representation of the electron-transfer triad based on crystal structures.
In our paper, we designed the RC maquette to be modular so that different donors, pigments, and acceptors could be added or removed without disrupting other binding sites. We showed that we could replace tyrosine with a cysteine-crosslinked ferrocene moiety and replace heme with a higher-potential iron porphyrin to create different permutations of DPA triads. We measured DPA triad quantum yields as high as 31% and charge separated state lifetimes as long as 350 ms. A DDPA tetrad of iron(II)-tyrosine-Zn porphyrin-heme demonstrated a 250 ms lifetime and 6.4% yield. These charge separation lifetimes are long enough to support chemical reactions as slow as the 1-2 ms turnover of water oxidation in Photosystem II7.
The modular RC maquette design allowed us to explore the properties of different redox triads and tetrads and will facilitate future extensions of the electron transport chain. We will focus future work on improving the thermodynamic efficiency and quantum yield as well as inserting catalytic sites with the goal of renewable fuel production.
1 Blankenship, R. E. et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 332, 805-809, (2011).
2 Umena, Y., Kawakami, K., Shen, J. R. & Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 angstrom. Nature 473, 55-U65, (2011).
3 Moser, C. C., Keske, J. M., Warncke, K., Farid, R. S. & Dutton, P. L. Nature of biological electron transfer. Nature 355, 796-802, (1992).
4 Faiella, M. et al. An artificial di-iron oxo-protein with phenol oxidase activity. Nat Chem Biol 5, 882-884, (2009).
5 Torres Martin de Rosales, R. et al. Spectroscopic and metal-binding properties of DF3: an artificial protein able to accommodate different metal ions. J Biol Inorg Chem 15, 717-728, (2010).
6 Huang, L. S., Cobessi, D., Tung, E. Y. & Berry, E. A. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc(1) complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. Journal of Molecular Biology 351, 573-597, (2005).
7 Vinyard, D. J., Ananyev, G. M. & Dismukes, G. C. Photosystem II: the reaction center of oxygenic photosynthesis. Annu Rev Biochem 82, 577-606, (2013).
8 Imahori, H. et al. Modulating charge separation and charge recombination dynamics in porphyrin-fullerene linked dyads and triads: Marcus-normal versus inverted region. J Am Chem Soc 123, 2607-2617, (2001).
My research focuses on protein design and photosynthesis.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Nice concept on Protein Maquettes on very challenging pathways. Super interesting!