DeepRTAlign: toward accurate retention time alignment for large cohort mass spectrometry data analysis
Published in Protocols & Methods, Cell & Molecular Biology, and Computational Sciences
Summary
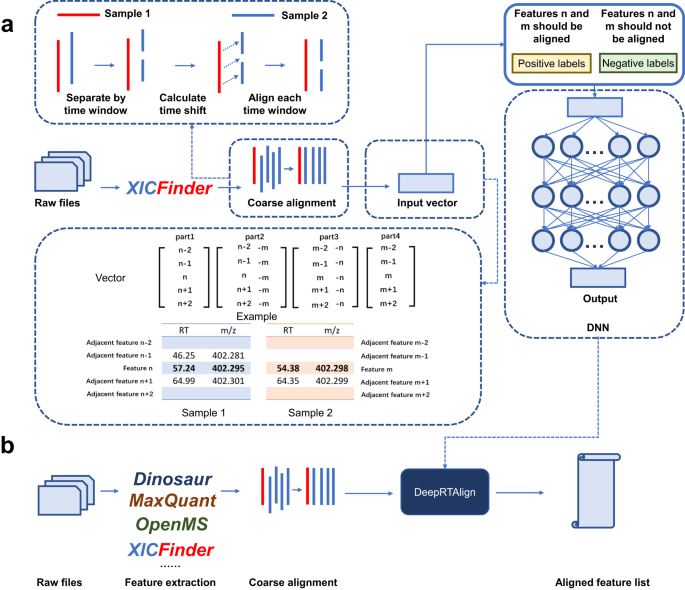
Retention time (RT) alignment is one of the crucial steps in liquid chromatography-mass spectrometry (LC-MS)-based proteomic and metabolomic experiments, especially for large cohort studies, and it can be achieved using computational methods; the most popular methods are the warping function method and the direct matching method. However, the existing tools can hardly handle monotonic and non-monotonic RT shifts simultaneously. To overcome this, we developed a deep learning-based RT alignment tool, DeepRTAlign1, for large cohort LC-MS data analysis. It first performs a coarse alignment by calculating the average time shift between any two samples and then uses RT and m/z as the main features to train its deep learning-based model. We demonstrate that DeepRTAlign has improved performances, especially when handling complex samples. Furthermore, using the MS features aligned by DeepRTAlign in a large cohort, we trained a classifier of 15 features to predict the early recurrence of hepatocellular carcinoma. The features were validated on an independent cohort using targeted proteomics with an AUC of 0.833. Being flexible and robust with four different feature extraction tools (feature lists from other tools can be used after conversing format to txt or csv files), DeepRTAlign provides an advanced solution to RT alignment in large cohort LC-MS data, which is currently one of the bottlenecks in proteomics and metabolomics research, especially for clinical applications.

Inspiration
The traditional MS-based biomarker discovery strategy mainly depends on the identification and quantification results of MS data, which has some inherent limitations, such as the low identification rate of MS spectra. At first, we aimed to develop a new strategy for biomarker discovery independent of the identification and quantification results of MS data, i.e., the so-called ID-free strategy. We detected and extracted potential precursors directly from MS spectra and applied a deep learning model to locate the differentially expressed precursor regions in a global way. Our previous work2 demonstrated the feasibility of this idea. In short, the whole workflow can be divided into three parts. The first part is data preprocessing which contains precursor detection and precursor filtering. The second part is training model using deep learning method to distinguish the tumor samples from the non-tumor ones. The third part is precursor selection using explainable artificial intelligence methods, such as Gradient-weighted Class Activation Mapping (Grad-CAM)3. However, the RT shifts across different samples significantly hindered accurate matching of target features to precursor ions for subsequent verification. So, we turned to looking for a suitable RT alignment tool to integrate into our ID-free strategy. We soon discovered that existing alignment tools were either already integrated into a certain pipeline, or only supported a specific feature extraction tool. Ultimately, we chose to create a highly versatile RT alignment tool that could seamlessly integrate with various feature extraction software. When designing DeepRTAlign, we added a coarse alignment step inspired by the warping function of existing tools. Then inspired by word embedding method in natural language processing, DeepRTAlign considers not only the feature to be aligned but also its adjacent features.
Future direction
While DeepRTAlign has shown some promising results, it is still under development and has some limitations that we are working to address:
- Dataset dependence: The current version of DeepRTAlign in our study is trained on a large-scale liver cancer dataset (HCC-T dataset)4. To improve its adaptability, we will provide a trainable version that allows users to train an ad hoc neural network using their own data.
- User-friendliness: Currently, DeepRTAlign is only available as a Python package, which can be cumbersome for some users. To improve accessibility, we plan to develop a user-friendly graphical interface.
- Processing speed: Although DeepRTAlign supports parallel processing, it may still be slow when processing large amounts of data. To address this limitation, we are exploring the possibility of implementing a C++ version for more efficient performance.
We are committed to improving DeepRTAlign and making it a valuable tool for the scientific community. We welcome feedback and suggestions from users, which will help us prioritize future development directions.
References:
1 Liu, Y. et al. DeepRTAlign: toward accurate retention time alignment for large cohort mass spectrometry data analysis. Nature communications 14, 8188, doi:10.1038/s41467-023-43909-5 (2023).
2 Dong, H. et al. A Deep Learning-Based Tumor Classifier Directly Using MS Raw Data. Proteomics, e1900344, doi:10.1002/pmic.201900344 (2020).
3 Selvaraju, R. R. et al. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. 2017 IEEE International Conference on Computer Vision (ICCV), 618-626, doi:10.1109/iccv.2017.74 (2017).
4 Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257-261, doi:10.1038/s41586-019-0987-8 (2019).
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in