Diagnostic impact of whole exome sequencing in neurometabolic disorders in Syrian children: a single center experience
Published in Genetics & Genomics, General & Internal Medicine, and Paediatrics, Reproductive Medicine & Geriatrics
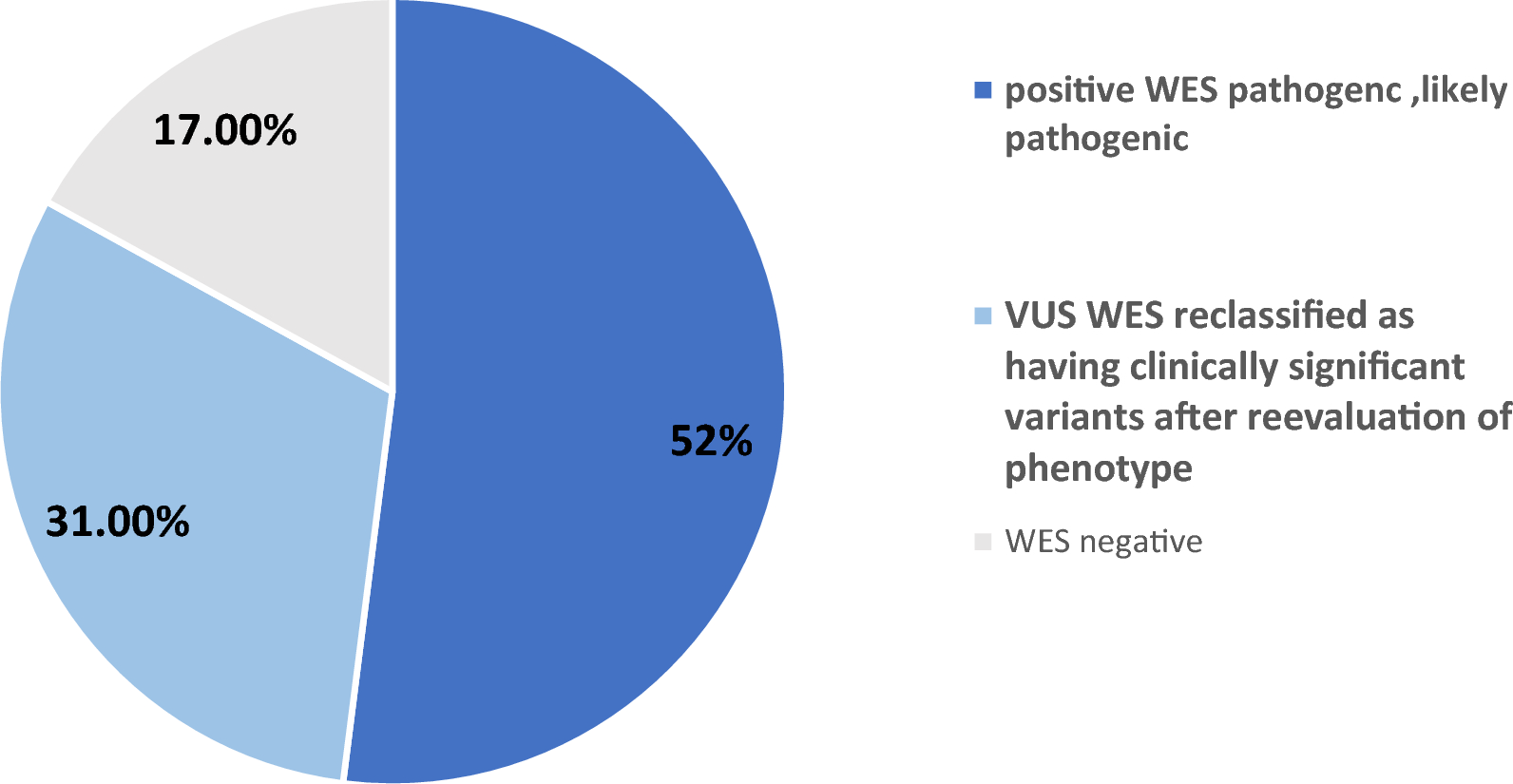

This study emerged from an urgent clinical need. We evaluated 54 children with unexplained neurological and metabolic symptoms at our center. Many of these patients had a history of consanguinity and had previously undergone long diagnostic journeys without clear answers. By applying WES, we identified pathogenic or likely pathogenic variants in 52% of cases and clinically relevant findings in 83%, including 14 novel mutations not previously reported.
Beyond diagnosis, WES had a direct impact on patient care:
Diagnostic reclassification occurred in 83% of cases
Treatment plans were adjusted in 74% of cases
Prognostic clarity and preventive counseling became possible for the majority of families
Our work highlights the transformative role of WES in diagnosing rare pediatric conditions, particularly in populations with high consanguinity and limited access to traditional genetic services. We hope this study encourages the integration of genomic diagnostics into neurology and pediatrics in similar settings worldwide.
We invite you to read our paper and join the conversation.
https://rdcu.be/ek5is
I am Dr. Rawan Al Khudari, a general pediatrician and clinical researcher with a strong academic record and deep commitment to advancing child health. I graduated top of my class and was honored for academic excellence. I also served as chief resident during my pediatric residency, where I developed leadership skills and mentored junior colleagues. My clinical and research interests span various pediatric fields, and I aspire to pursue a subspecialty in pediatric cardiology. I am passionate about evidence-based care, academic growth, and contributing meaningfully to the global pediatric community.
Follow the Topic
-
Orphanet Journal of Rare Diseases
An open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs and publishes high-quality reviews on specific rare diseases.

Related Collections
With Collections, you can get published faster and increase your visibility.
The Emerging Frontiers of Genetic Screening in Inherited Disorders
The core challenge of modern preventive medicine is the accurate and timely identification of individuals at risk for genetic disorders. For decades, genetic testing has been gated by restrictive clinical criteria and "phenotype-first" models, which often rely on family history or specific symptoms to trigger a diagnostic workup. However, emerging evidence highlights a significant diagnostic gap: a substantial proportion of individuals carrying clinically actionable pathogenic variants are currently missed because they do not meet traditional testing guidelines or lack a documented family history of disease.
This Collection focuses on how the next generation of screening technologies can expand diagnostic capacity and maximize diagnostic yield. We are moving toward a "genome-first" era, in which criteria-independent testing and the integration of multimodal data enable the identification of individuals at risk who were previously undetected within the healthcare system. By leveraging advanced sequencing technologies and computational infrastructures, we can shift from reactive diagnostics to proactive, large-scale screening strategies that improve clinical outcomes across multiple medical disciplines.
Key Areas of Interest
We invite original research, reviews, and clinical perspectives that explore the technological and clinical pathways to increasing diagnostic sensitivity and specificity:
- Expanding the Diagnostic Net: Evaluating the clinical utility of criteria-independent or universal screening to identify high-risk individuals who would otherwise remain undetected under standard guidelines.
- Genome-First Implementation: Analysing the impact of population-based genomic screening in uncovering hereditary diseases or pathogenic variants associated with inherited diseases before the onset of clinical symptoms.
- Synergistic Diagnostic Modalities: The integration of DNA sequencing with other high-resolution markers, such as biochemical profiling or functional assays, to refine risk assessment and increase diagnostic precision.
- Opportunistic Screening and Secondary Findings: Best practices for managing and reporting actionable genetic findings discovered during routine clinical sequencing.
- Scalable Screening Architectures: Innovative approaches to integrating large-scale genetic data into healthcare systems, including the use of electronic health records to support automated risk identification.
- Overcoming Implementation Barriers: Strategies to mitigate disparities in access to screening, focusing on populations in which reliable family history information is limited.
All submissions in this Collection undergo the journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief. As an open access publication, this journal levies an article processing fee (details here). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief.
Publishing Model: Open Access
Deadline: Jan 16, 2027
European Reference Networks: Advancing Collaboration and Care for Rare and Complex Diseases
This Collection highlights the core activities and strategic impact of the European Reference Networks (ERNs) in addressing the challenges of rare and complex diseases. Contributions will explore the ERNs' roles in cross-border healthcare, clinical expertise sharing, care pathway development, patient registry advancement, training, and research coordination. Emphasis is placed on collaborative models, digital tools, and patient-centered approaches that are transforming diagnosis, care, and innovation across Europe.
Please note that this Collection accepts submissions ONLY from authors invited by the Guest Editors.
All submissions in this collection undergo the journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief. As an open access publication, this journal levies an article processing fee (details here). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief.
Publishing Model: Open Access
Deadline: Dec 31, 2026