Introduction
The genomes of any two people are generally said to be >99% identical [1], but it’s the differences between them, known as variants, that are critical. Not only do they underpin a person’s uniqueness but they can have a direct influence on physical characteristics – including heritable diseases and cancer. ‘Variant calling’, an umbrella term for a large number of computational methods for detecting variants, is therefore a vitally important task. To identify (‘call’) the variants in a given sample, its DNA is sequenced and compared with a standard human ‘reference’ genome, resulting in a set of variants.
There are now many variant calling methods exceptionally accurate at calling single nucleotide variants, those where only one base differs from the reference. However, these represent only one category of variant, a relatively simple one. We were interested in our study with structural variants, a broader category far more challenging to call accurately yet increasingly associated with many complex phenotypes, including autism and schizophrenia [2]. Structural variants are often either missed or misinterpreted by variant calling software. This is because of their complexity: they can comprise long stretches of DNA which relative to the reference genome are either deleted, inverted, duplicated, or inserted – or any combination of these things.
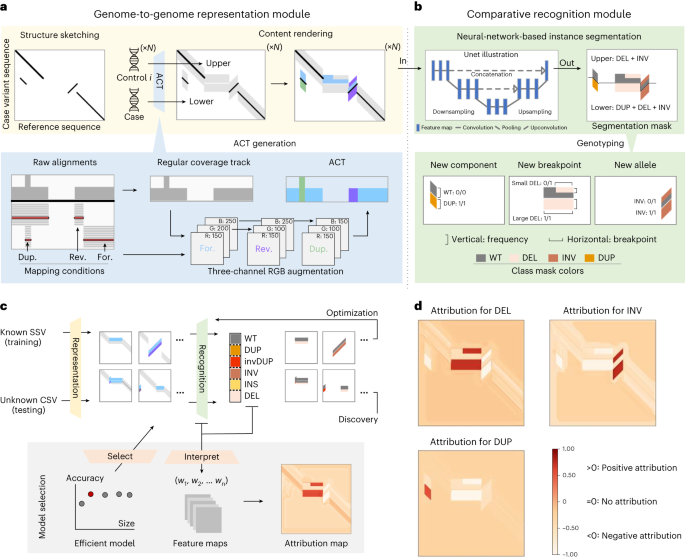
Due to their length, it’s only practically possible to call the most complex structural variants using long-read sequencing technologies, such as Oxford Nanopore or Pacific Biosciences. To that end, we have previously published SVision [3], structural variant calling software designed for long-read data. A common means of interpreting long-read data is by first ‘aligning’ the reads to the reference genome, identifying the most likely location in the genome from which they have arisen. SVision works by changing the way these alignments are ‘seen’, computationally. DNA sequence alignments are in effect paired strings of DNA ‘text’. SVision encodes this text in the form of an image instead, in effect adapting the problem of variant calling (“what are the points of similarity and difference in an alignment?”) to one amenable to deep-learning algorithms, the current state-of-the-art.
It’s this process of ‘encoding’ which ultimately determines what the algorithm can do – the more data you can encode into an image, the greater the variety of variant calling tasks you can tackle. However, SVision, while a powerful tool, was limited in that it could only call variants for one sample at a time. Our aim with SVision-pro was to expand the capabilities of SVision, encoding more information into its images, thereby allowing the algorithm to handle multiple samples (that is, sets of alignments) simultaneously. This would allow you to identify structural variants that differ not just between a given genome and the standard ‘human reference’ but between any sets of genomes you choose.
But why would you want to do this?
There are two main reasons to want to call variants between one or more specified genomes, rather than between any given genome and an external ‘reference’. Firstly, it would let you directly compare the genome of a child with that of their parents. This is important because de novo variants – those present in a child but not the parents – make a particularly significant contribution to human disease, globally affecting approximately 1 in 300 new births. In this case, you’d be able to call those variants associated with an unexpected (no family history) disorder in a child. Secondly, it would let you directly compare the genome of a tumour with that of healthy cells from the same person. In this case, you would be able to call those variants associated with the progression of cancer. With SVision-pro, our aim was to facilitate the accurate discovery of variants in these particular cases.
What next for structural variant calling?
A complete catalogue of genetic variation is a long-term goal of human genome research, with structural variants one of its last frontiers. A recent study of 405 unrelated Chinese individuals, for instance, identified 60,000 novel structural variants, ones not already present in the human reference genome [4]. Indeed, in recognition of the fact that ‘one genome is not enough’ – one reference genome cannot capture the full extent of human genome diversity [5] – large research consortiums are actively developing ‘pangenome’ references instead, condensing the data from a diverse spread of individuals into improved resources. Last year, a draft produced by the Human Pangenome Reference Consortium [6], along with Chinese [7] and Arab [8] pangenomes, have all been reported.
The landscape of structural variation in human populations is evidently a rugged one, underexplored in comparison and challenging to navigate. We can think of it as the terra incognita of the genome. Our aim with SVision-pro was to further improve discoveries in this area, but there is always scope to go further. Returning to the concept of ‘encoding’, if more information could be added to the image, it’s possible we could start to call and compare variants not only between one or more specified genomes but hundreds or thousands at a time; that is, to call structural variants across entire populations. Our ultimate vision here is both a hopeful and a global one: to contribute to surveying the spectrum of human variation in its entirety.
References
1. https://www.genome.gov/about-genomics/educational-resources/fact-sheets/human-genomic-variation
2. Weischenfeldt, et al. (2013) Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics.14:125-138 (2013). https://doi.org/10.1038/nrg3373.
3. Lin, et al. (2022) SVision: a deep learning approach to resolve complex structural variants. Nature Methods. 19:1230-1233. https://doi.org/10.1038/s41592-022-01609-w.
4. Wu, et al. (2021) Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation. Nature Communications. 12:6501. https://doi.org/10.1038/s41467-021-26856-x.
5. Yang, et al. (2019) One reference genome is not enough. Genome Biology. 20(1):104. https://doi.org/10.1186/s13059-019-1717-0.
6. Liao, et al. (2023) A draft human pangenome reference. Nature. 617:312-324. https://doi.org/10.1038/s41586-023-05896-x.
7. Gao, et al. (2023) A pangenome reference of 36 Chinese populations. Nature. 619:112-121. https://doi. org/10.1038/s41586-023-06173-7.
8. Uddin, et al. (2023) A draft Arab pangenome reference. ResearchSquare pre-print. https://doi.org/10.21203/rs.3.rs-3490341/v1.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in