DNA methylation landscapes of 1538 breast cancers reveal a replication-linked clock, epigenomic instability and cis-regulation
Published in Cancer


Project motivation
Breast cancer is one of the leading causes of cancer death in women and is a heterogeneous disease displaying distinct clinical courses and therapeutic responses. Accordingly, the scientific community has strived to catalogue and characterize the molecular drivers in breast cancer to bring us closer towards the goal of personalized medicine [Cancer Genome Atlas Network, 2012]. I joined Carlos Caldas’ laboratory at the Cancer Research UK Cambridge Institute as a doctoral researcher to support this aim. It was a perfect time to join since the laboratory (in collaboration with the University of British Columbia) had recently established the METABRIC consortium dataset which represents a large cohort of 2000 primary breast tumors with detailed clinical histories. Efforts from the lab so far, had focused on high-throughput genomic and transcriptomic profiling of the METABRIC cohort [Curtis et al., 2012; Pereira et al., 2016; Rueda et al., 2020], and revealed that breast cancer subtypes possess distinct molecular wiring that are dominated by copy number aberrations, with a few genes (such as TP53 and PIK3CA) hit by somatic mutations.
However, in addition to genetic alterations, the de-regulation of cancer cells is also driven by a complex interplay of multiple layers of aberrant epigenetic mechanisms. Of these, DNA methylation remains the most extensively characterized, and the cancer methylation landscape has been analyzed in many tumor types, mostly using microarrays. CpG island hypermethylation has been identified in thousands of genes in breast cancer and linked with gene repression, and global methylation loss has also been observed as a cancer hallmark [Zhou et al. 2018]. However, the forces that drive these pervasive methylation changes in tumors, as well as the precise consequences of methylation aberrations on tumorigenesis are still poorly understood.
Study development
From this perspective, the breadth and scope of the METABRIC study along with the availability of multi-dimensional genomic and transcriptomic profiles for the cohort, provides a vital resource to investigate the epigenomic landscape in breast cancer. We profiled DNA methylation across 1538 breast tumors (1179 ER+ and 350 ER- tumors) and 244 normal breast tissues using reduced representation bisulfite sequencing (RRBS). Once the data was ready to analyze, we teamed with Amos Tanay’s Lab at the Weizmann Institute of Science to determine the multi-factorial processes responsible for inducing cancer DNA methylation alterations and provide an understanding of its potential driver role in breast cancer.
Study findings
Analyzing genome-wide cancer methylation profiles is challenging since the readouts are a consequence of the convergence of multiple mechanisms, dynamics and biases. In order to unravel this complexity, we developed a comprehensive and systematic strategy called Methylayer that places each regulatory layer in the context of other mechanisms. The principle underlying Methylayer relies on integration of gene expression, genetics and clinical information for i) computational peeling of the confounding effects of the tumor microenvironment (TME); ii) the inference of global trends that can stochastically affect a majority of the methylome; and finally, iii) screening for potential driver candidates with evidence of epigenetic cis-regulation based on this top-down approach. Furthermore, for each of these layers, we investigated how these tumor mechanisms affect clinical outcomes and prognosis.
Tumor microenvironment (TME)
Tumor tissues consist of not only cancer cells but also variable populations of resident and infiltrating cell types [Ali et al., 2020], known as TME, that can act as major confounders influencing methylation signals obtained from tumor biopsies. Methylayer utilizes an unsupervised approach relying on the integration of gene expression with promoter methylation data to identify TME signatures. We detected i) a strong immune signature anchored by expression profiles of many marker genes (CD3, CD8) and checkpoints (CTLA4, PD-1); and ii) a cancer associated fibroblast (CAF) signature was anchored by stromal markers such as FAP, CAV1 and VIM. To facilitate robust deconvolution of these TME effects, we applied a novel K-nn normalization algorithm that provided Methylayer substantially reduced TME bias for delineating tumor methylation layers downstream.
Global remodeling factors
Next, Methylayer identified broad trends of methylation aberration in the breast tumors using clustering of the TME-normalized methylation that yielded highly correlated groups of CpG sites. We identified a cancer signature demonstrating reduced methylation levels in breast tumors (compared to almost fully methylated in the normal tissues) that could be explained by an accumulation of methylation errors strongly correlated with genome replication trends. This was denoted as the clock layer and is comprised of regions characterized by low CpG content and under-represented at regulatory elements including enhancers. However, the clock signature was not associated with transcriptional programs (except for a few cancer testis antigens, CTA) or tumor aggressiveness.
We also identified two further global methylation signatures – the first was defined by methylation gain in breast tumors in regions that were unmethylated in the normal controls (MG layer); and the second defined by regions that are partially methylated in normal tissues but show a spectrum of reduced methylation in tumors (ML layer). Both signatures were characterized by high intra-tumor methylation heterogeneity indicating that they were the outcome of multiple stochastic events rather than takeover of specific epi-alleles, and hence we termed as epigenomic instability signatures. In sharp contrast to the loss clock layer, the two epigenomic instability signatures were enriched in promoters and enhancers, and associated with tumor gene expression programs (such as cell cycle and embryonic development) as well as tumor progression.
Finally, we also identified a cluster of X-linked promoters whose methylation profiles provided a powerful dosage compensation mechanism associated with X chromosome inactivation.
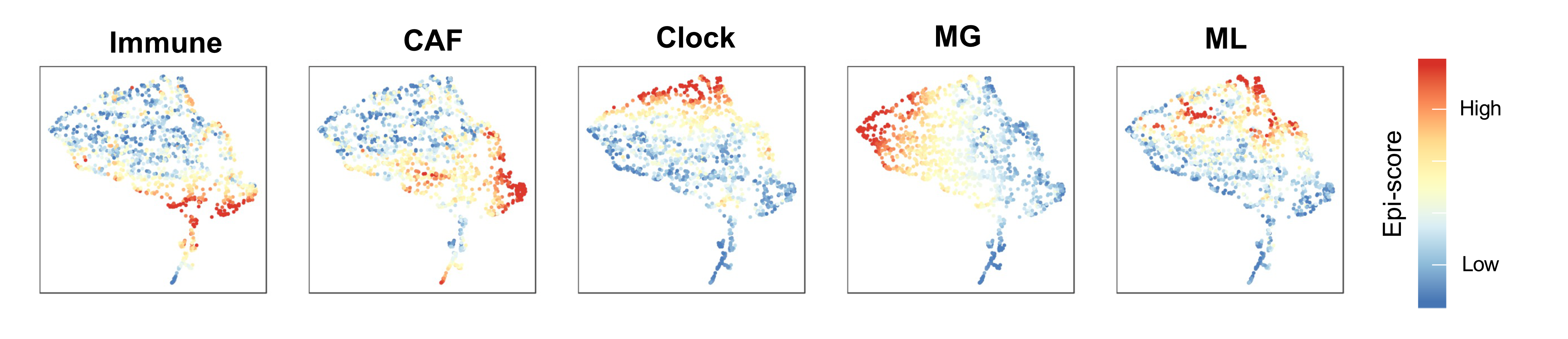
Blog Figure 1: Projection of METABRIC tumor samples on a unified epigenetic signatures space, colored by the 5 epigenetic scores.
Link with genomic aberrations and prognosis
The five epigenomic signatures (Immune, CAF, clock, MG and ML) converge to explain the global methylation landscape in breast cancer (Blog Figure 1). We showed that these epigenetic signatures, and in particular MG and ML epigenetic instability are correlated with genomic features of tumors including key somatic mutations such as TP53, PIK3CA and CDH1. Both MG and ML signatures were also predictive of higher tumor grade and poor breast cancer-specific survival (BCSS) even after considering clinical, genetic and transcriptional metrics (Blog Figure 2).
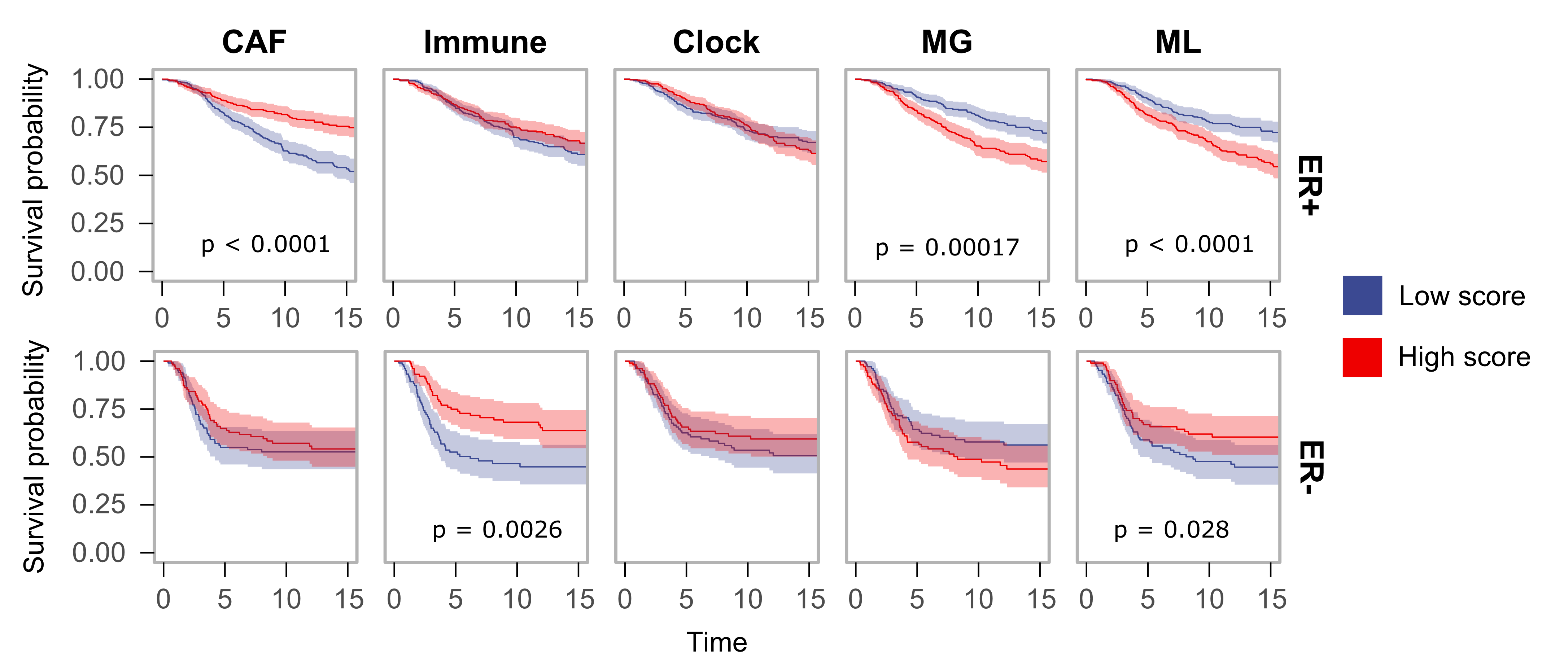
Blog Figure 2: Kaplan-Meier survival plots for ER+ (top, n = 1108) and ER- (bottom, n = 310) tumors grouped into high-scoring and low-scoring groups for each epigenomic signature (top 1/3 and bottom 1/3 of the samples). 95% confidence intervals are shown. Log-rank p-values for survival differences are reported.
In cis regulatory role of methylation alterations
Each of the broad trends of methylation aberration defined above are likely to be a consequence of the carcinogenic process. A major challenge in identifying methylation alterations that may have a direct regulatory role on tumor phenotypes (i.e. a driver event) is to discriminate those that are part of global remodeling in-trans trends versus those loci that are affected by methylation changes in-cis. We reasoned that specific methylation-expression in cis regulatory relationships can be supported when the methylation level (at a promoter or potential enhancer) correlates with its target gene expression in-cis at significantly higher levels than those predicted by the global remodeling signatures working in-trans. Using this screening approach, Methylayer identified hundreds of distinct regulatory promoters as well as thousands of distal cis elements in breast cancer that may act as drivers of inter-tumor expression variation for individual genes. Key examples of cis-regulated promoter hypermethylation effect include the classical BRCA1 silencing in ER- tumors, and also novel examples such as KRT7 repression in ER+ tumors.
Remarkably, the selected promoters exhibited significantly reduced methylation heterogeneity, suggestive of epigenetic convergence, or even selection at these loci during tumorigenesis.
Summary
Our layered analysis of tumor methylation dynamics in 1538 breast tumors provided a unified model delineating the multi-factorial processes giving rise to breast cancer DNA methylation.
The model represents 6 global trends that affect breast DNA methylation profiles, two involving TME effects of immune and stromal cells, one representing replication-linked hypomethylation clock, one involving X chromosome dosage compensation and two representing epigenetic instability (in trans methylation gain and loss) at enhancers and promoters. We also demonstrated methylation in hundreds of promoters and thousands of distal elements to be correlated with gene expression specifically in-cis, including the classical BRCA1 hypermethylation effect. This may suggest epigenomic instability predisposes tumors to greater regulatory variation and flexibility, in a way resembling the impact of genomic instability on tumors. The discovery that epigenomic instability is pervasively observed in high grade tumors, with prognostic power that is synergistic to clinical and genetic markers, may further hint toward its possible functional impact.
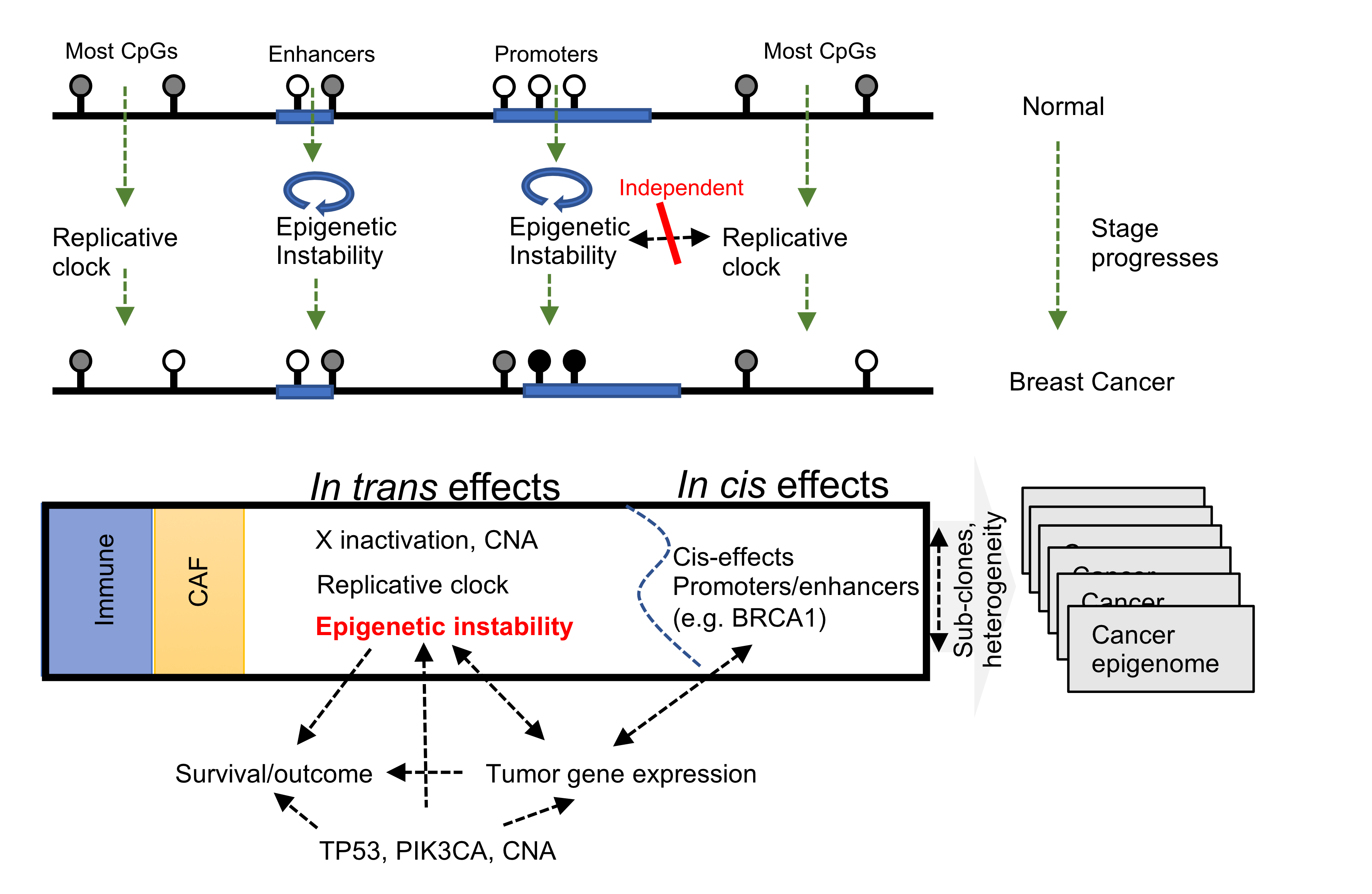
Blog Figure 3: Unified model delineating the multi-factorial processes giving rise to breast cancer DNA methylation.
Blog contributors: Rajbir Nath Batra, Aviezer Lifshitz, Amos Tanay and Carlos Caldas
Link to the paper: https://www.nature.com/articles/s41467-021-25661-w
References
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). https://doi.org/10.1038/nature11412
Curtis, C., Shah, S., Chin, SF. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). https://doi.org/10.1038/nature10983
Pereira, B., Chin, SF., Rueda, O. et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat Commun 7, 11479 (2016). https://doi.org/10.1038/ncomms11479
Rueda, O.M., Sammut, SJ., Seoane, J.A. et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404 (2019). https://doi.org/10.1038/s41586-019-1007-8
Zhou, W., Dinh, H.Q., Ramjan, Z. et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 50, 591–602 (2018). https://doi.org/10.1038/s41588-018-0073-4
Ali, H.R., Jackson, H.W., Zanotelli, V.R.T. et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat Cancer 1, 163–175 (2020). https://doi.org/10.1038/s43018-020-0026-6
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in