Dysfunctional microglial mitochondria impair brain development
We show for the first time that the mitochondrial activity of microglia is essential for postnatal brain development, suggesting a direct microglial contribution to primary mitochondrial diseases and pointing to mitochondria as a new pathway to modulate microglial function.
The brain is a fundamental organ that determines who we are and our ability to interpret and interact with the outside world. Countless studies address the organization and physiological mechanisms of the brain, which we are still very far from fully understand. An interesting and important question is how the brain is formed, reaching its state of maturity and functionality in each individual. In general terms, developmental biology is full of complex and delicate processes, which require the coordinated activity of different types of cells in time and space, a process that has fascinated many scientists (including those in our laboratory) and that is especially remarkable in the case of the brain, given its incredible level of complexity [1].
Microglia and neurological diseases
Traditionally in neuroscience most studies used to focus exclusively on neurons, since they are the structural and functional units of the nervous system. At present, the effort of the scientific community has diversified and includes glial cells or neuroglia, a group of cells for which we are attending an explosion of works identifying multiple and essential functions [2]. At this point, nobody doubts that glial cells are essential to support (physically and metabolically) and modulate the function of neuronal cells. Among the different types of glial cells, we and other laboratories are particularly interested in microglia, the brain's innate immune system. These cells are fundamental to brain development, acting as “sweepers” to eliminate neurons and non-functional connections, finely sculpting the brain's functionality based on the postnatal experience. The study of microglia has gained great importance in recent years due to the close relationship between the activity of these cells and different diseases of the nervous system [3]. For example, different laboratories have demonstrated a key role of microglia in the progression of Alzheimer's disease. In this context, our group recently described that microglia depend on oxygen and the activity of mitochondria, the cellular powerhouses, and that Alzheimer's disease affects microglial activity through these pathways [4].
New data
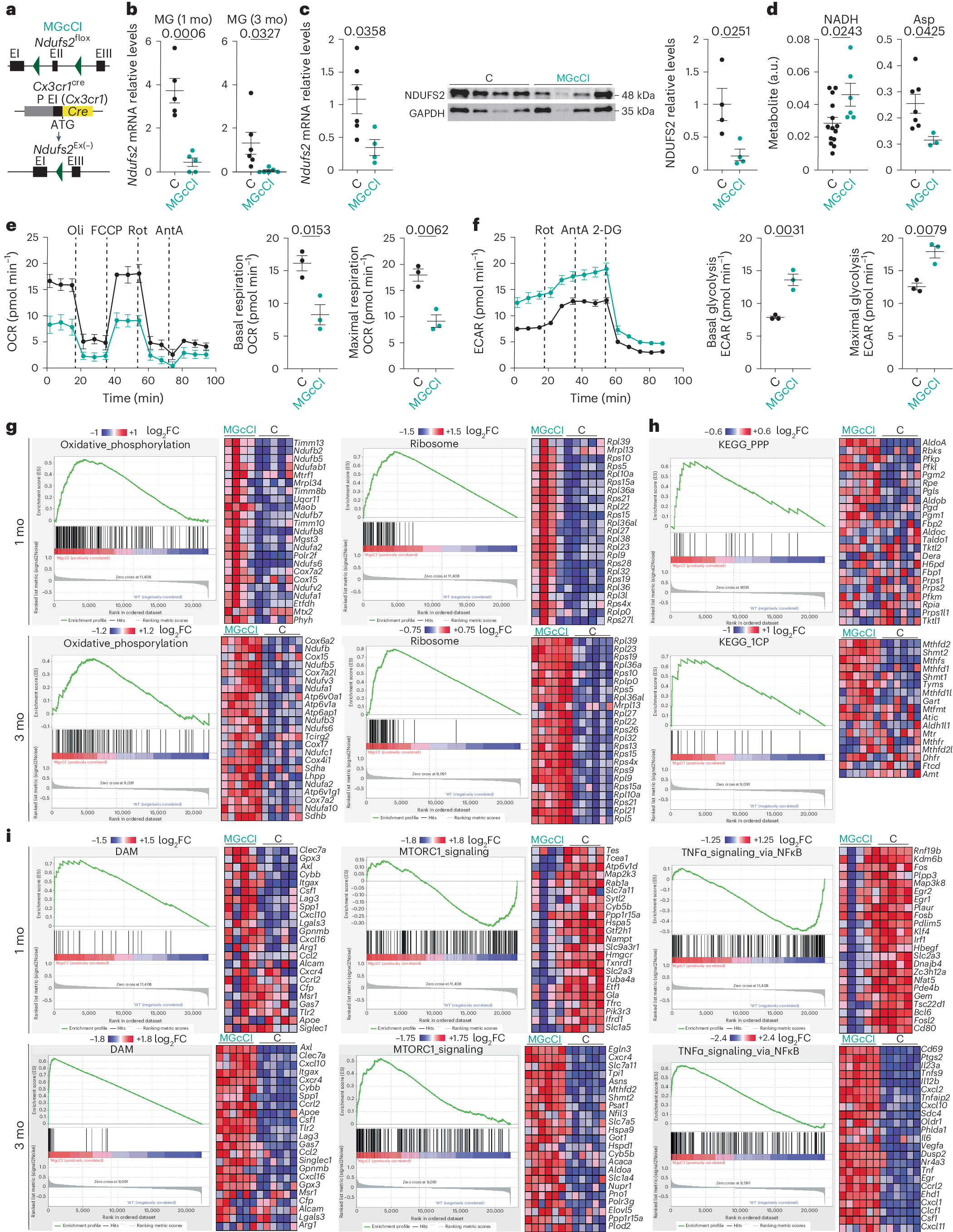
After the publication mentioned above [4], we came to a surprising (by then) conclusion: microglia activity promotes mitochondrial metabolism. We knew this was true during neurodegeneration for both human and mouse models, what drove us to study the contribution of mitochondria to the activity of microglia. To do this, we used genetic techniques and mouse models to reduce, specifically in microglia, the activity of one of the central elements that allows the use of oxygen in the mitochondria, the mitochondrial complex I. Disrupting in this way the microglial mitochondria from the embryonic stage did not prevent normal birth of the animals, and surprisingly, the activity of this mutated microglia at 1-month-old mice was at least as high as in healthy conditions. In some way, these cells were overcoming the loss of mitochondrial functionality during brain development, a moment at which they are known to be particularly active. This might indicate that we were mistaken, and that microglial activity did not depend on mitochondria. However, a new publication was released, showing for the first time the upregulation of microglial mitochondria during brain development [5] (until then, as mentioned above, this had only been observed in pathology-associated microglial activity). In addition to this data supporting our hypothesis, we had observed by then that at 3 months old animals, CI-deficient microglia showed reduced ability to perform their normal function, present altered morphology (Fig. 1), and express a group of genes that microglia express in Alzheimer's disease and other neurodegenerative disorders. This evidence suggests that the loss of mitochondrial complex I in microglia drive the cells to a pathological state, and importantly, has an impact on the brain, since we detected alteration of neurons and other glial cells. As a result, the animals carrying CI-deficient microglia showed cognitive decline (Fig. 2) and premature death. The latter phenotype was a big surprise for us, given that previous studies had shown that the absence of microglia in mice does not produce any evident brain defect after birth, and animals can live normally for at least 9 months [6]. Therefore, an important conclusion is that having dysfunctional microglia is much more detrimental for brain development than lacking microglia.
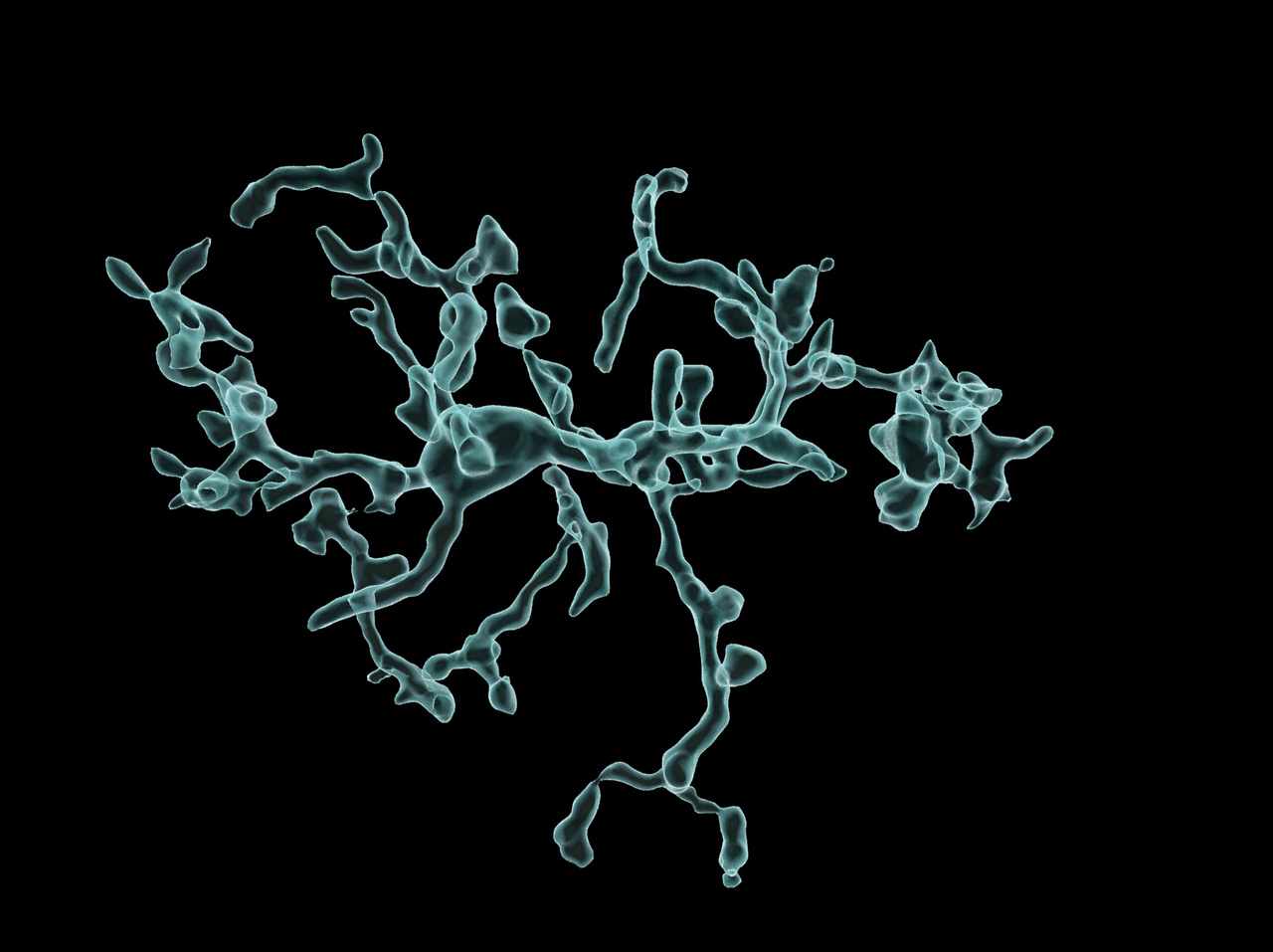
3D reconstruction of a representative cell from control (WT) and CI-deficient (Mutant) microglia.
Adapted from Mora-Romero et al., Nat Metab 2024.
Behavior of control (WT) and CI-deficient (Mutant) microglia mice in an open field maze. Adapted from Mora-Romero et al., Nat Metab 2024.
Relevance of the study
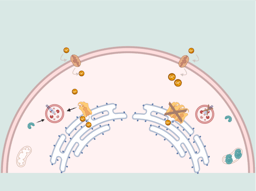
Mutations similar to the one we have made in our mouse models are associated in humans with Leigh syndrome, a primary mitochondrial disease that affects 1 in 40,000 births worldwide. This syndrome causes neurological problems and previous studies have defined the relevance of microglia in the progression of the disease [7]. Our study goes one step further and points to microglia as a cell directly responsible for the disease, contributing to its progression not only due to the damage that can occur in other cells such as neurons, but also because it can alter brain development (Fig. 3). Therefore, this study opens new therapeutic targets and defines the windows of action in primary mitochondrial diseases. Furthermore, the results may have consequences for how we interpret the neuroinflammation underlying neurodegeneration processes and how microglia activity could be controlled in these processes. This will be the subject of future studies.
Diagram summarizing the main result of the study. Created with BioRender.com
[1] Piwecka, M., Rajewsky, N. & Rybak-Wolf, A. Single-cell and spatial transcriptomics: deciphering brain complexity in health and disease. Nat Rev Neurol 19, 346–362 (2023). https://doi.org/10.1038/s41582-023-00809-y.
[2] Mohapatra, S., Mukherjee, S., Swaroop, S. & Basu, A. The Biology of Glial Cells: Recent Advances (eds. Patro, I., Seth, P., Patro, N. & Tandon, P.N.). 459–505 (Springer Singapore, 2022).
[4] March-Diaz, R., Lara-Ureña, N., Romero-Molina, C., Heras-Garvin, A., et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nat Aging 1, 385–399 (2021). https://doi.org/10.1038/s43587-021-00054-2.
[5] He, D., Xu, H., Zhang, H., Tang, R., et al. Disruption of the IL-33-ST2-AKT signaling axis impairs neurodevelopment by inhibiting microglial metabolic adaptation and phagocytic function. Immunity, 11;55(1), 159-173.e9 (2022). https://doi.org/10.1016/j.immuni.2021.12.001.
[6] Rojo, R., Raper, A., Ozdemir, D.D., Lefevre, L., et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun 10, 3215 (2019). https://doi.org/10.1038/s41467-019-11053-8.
[7] Daneshgar, N., Leidinger, M.R., Le, S., Hefti, M., et al. (2023) Activated microglia and neuroinflammation as a pathogenic mechanism in Leigh syndrome. Front. Neurosci, 16:1068498 (2023). https://doi.org/10.3389/fnins.2022.1068498.
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
This journal publishes work from across all fields of metabolism research that significantly advances our understanding of metabolic and homeostatic processes in a cellular or broader physiological context, from fundamental cell biology to basic biomedical and translational research.
We use cookies to ensure the functionality of our website, to personalize content and advertising, to provide social media features, and to analyze our traffic. If you allow us to do so, we also inform our social media, advertising and analysis partners about your use of our website. You can decide for yourself which categories you want to deny or allow. Please note that based on your settings not all functionalities of the site are available.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in