Encoding NMR chemical shifts into a biophysical index
Published in Chemistry
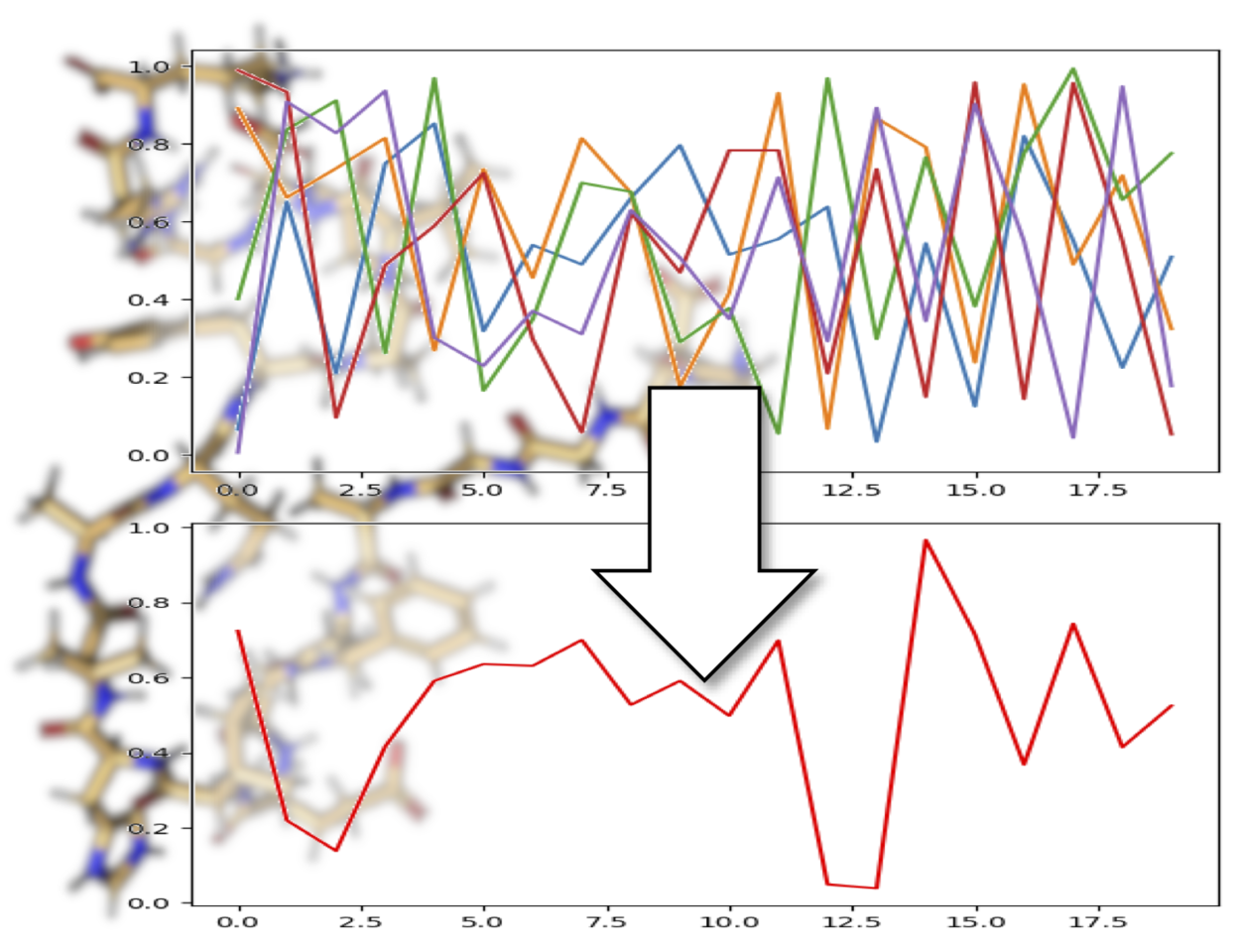
Chemical shifts are the bread and butter of Nuclear Magnetic Resonance (NMR) measurements. They represent the frequencies at which atoms resonate in NMR spectrometers, and so reflect the (chemical) environment of those atoms. For proteins, assigning the chemical shifts to the atoms of their amino acid residues is the first step towards their NMR-based interpretation. Once assigned, these chemical shifts contain a wealth of information about the behavior of the protein in solution. They can indicate the conformation of each amino acid1, and even their dynamics2. Such methods use reference chemical shift values to pinpoint the ‘default’ state of each amino acid type, so enabling different amino acids to be compared to each other.
While working on a project in the lab to estimate the behavior of amino acids from their chemical shift values, we continued to run into a key problem: which set of reference chemical shift values should we use? These values are experimentally determined from short peptides in solution, where the central amino acid is assumed to be in a conformationally neutral state. Different sets have different chemical shift values, however, indicating that these peptides in fact do have inherent conformational preferences. Because we do not know the actual conformations these peptides adopt, it is also impossible to know which of these sets is the ‘best’ (most default). A second challenge we encountered is that chemical shift values for the atoms within one amino acid are interconnected; they change in concert in the hyper-dimensional space they occupy, typically consisting of at least 6 dimensions (atoms). Model-based estimation methods cannot take this into account, and human minds are not able to grasp more than 3 dimensions. Artificial intelligences on the other hand do not have such limitations: the space in which neural networks operate has an arbitrary number of dimensions.

This led us to try and interpret the chemical shift values in their native space, without any standardizations or assumptions about what the values themselves mean. With ShiftCrypt, we “ask” a neural network based autoencoder to “understand” chemical shifts for us and to summarize them in a single dimension (Figure 1). The only assumption we make is that changes in hyper-dimensional shift space have a similar meaning for each amino acid type. In other words, we assume the ShiftCrypt value reflects the overall conformational and dynamic nature of an amino acid, regardless of its type.
Despite this hands-off approach, ShiftCrypt is able to capture typical protein-structure based information such as secondary structure content, without being limited to one particular type of information. The short ‘default state’ peptides, for example, turn out to have quite different and unexpected biophysical biases when viewed the ShiftCrypt way. For full proteins, ShiftCrypt can compare their biophysical characteristics directly, even when they have very different amino acid sequences.
We think ShiftCrypt is a nice illustration of how artificial intelligence, when applied correctly, can shed new light on existing data. Man-made models are, and will continue to be, very useful, especially in case few data are available. Such models, however, always incorporate our current thinking and preconceptions about the interpretation of the data. Artificial intelligence methods do not have this restriction, and given sufficient unbiased data, they can help us to develop new understanding.
1. Wishart, D. S., Sykes, B. D. & Richards, F. M. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry31,1647–1651 (1992).
2. Berjanskii, M. V. & Wishart, D. S. A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc.127,14970–14971 (2005).
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in