Finding “chemical antibodies” is no longer like finding a needle in a haystack
Published in Bioengineering & Biotechnology
Researchers have to find an aptamer from a library containing 1015 candidates. This number is 100,000 times more than the population of the world. Traditionally, billions of target molecules are mixed with the library in a solution. After reaching an equilibrium, the solution is filtered through a membrane with the hope that the complexes of target molecules and specific aptamers will be concentrated on the membrane with undesired aptamer candidates removed. Unfortunately, molecular binding is a physical interaction. During the filtration step, specific candidates can fall apart from target molecules, go through the membrane, and get lost. Moreover, the candidates can go together with target molecules through the membrane. Most importantly, the library may be concentrated on the membrane because they can also bind the membrane in addition to target molecules. Therefore, researchers have to repeat this procedure. Before doing so, the molecules on the membrane will be amplified to generate a new aptamer library while many of them bind the membrane rather than target molecules. This procedure will be repeated ten to fifteen times in general. However, amplification is based on polymerase chain reaction (PCR) that has a bias to amplify some candidates instead of treating all candidates equally. Researchers have found that this bias can become more and more severe with the increase of repeating times. As a result, while researchers spend a huge amount of time repeating the procedure of looking for aptamers from the library, they may not get what they want eventually. This time-consuming procedure is accompanied by intensive labor and high costs without guaranteed success. Therefore, the procedure of finding aptamers is frustrating to not only beginners but also experienced researchers, like finding a needle in a haystack.
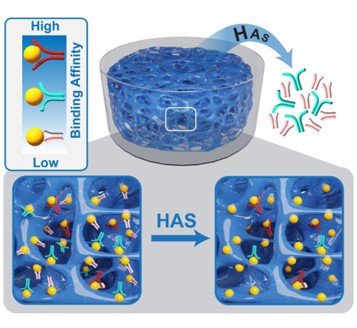
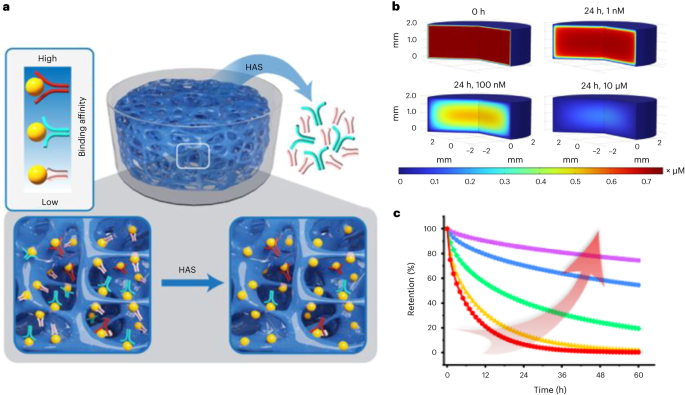
To make the procedure of aptamer selection easy and efficient, researchers have developed many elegant methods such as microbead-based selection, capillary electrophoresis-based selection, and microfluidic-based selection. However, these methods rely on instruments that can be too complicated for many researchers without specific trainings. More importantly, challenges such as PCR amplification bias, conformational changes in proteins or aptamers, low aptamer diversity, and particularly nonspecific binding to the selection matrix and target molecules may still exist. Therefore, we created a new method for aptamer selection with a different principle, i.e., a coupled binding-diffusion process in a three-dimensional, non-fouling, and macroporous space. This method was named HAS (i.e., hydrogel for aptamer selection).
Ideally, a defined three-dimensional permeable space only has target molecules and the library. All target molecules are magically immobilized whereas the library is free. Thus, all candidates in the library can freely leave this defined space if they do not bind the immobilized target molecules with a high strength. Conversely, the candidates will be imprisoned in this space if captured tightly by the immobilized molecules. Moreover, a desired aptamer candidate will re-bind an immobilized molecule if it falls apart from a formerly bound target molecule. Therefore, this candidate will not be easily lost during the procedure of diffusion. Under this ideal situation, aptamer selection can be achieved in one single step without the need of a reiterative process. All concerns with reiterative operations are avoided.
We created a pseudo-magic space using a non-fouling molecule, polyethylene glycol (PEG) since a non-fouling material would allow for minimal non-specific binding of the library. The data show that the library diffused out of the macroporous PEG hydrogel very quickly. By contrast, once the PEG hydrogel was functionalized with target molecules, the library’s apparent diffusion was slowed down. An aptamer enrichment pool was acquired in a single step. It holds true to all five protein targets that represents negatively, neutrally, and positively charged molecules with a large range of isoelectric points. Based on the next generation sequencing, we were able to identify at least one high-affinity aptamer for a target molecule.
In the future, we will apply or tune this method to do aptamer selection against non-protein targets.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in