Finding the needle in the haystack: translating big omics data into actionable clinical biomarkers
Published in Chemistry, Protocols & Methods, and Genetics & Genomics

Explore the Research
Discovery of sparse, reliable omic biomarkers with Stabl - Nature Biotechnology
Stabl selects sparse and reliable biomarker candidates from predictive models.
By Julien Hedou, Ivana Maric, Jakob Einhaus, Gregoire Bellan, Dyani Gaudilliere, Brice Gaudilliere, on behalf of all authors:
At the intersection of biology and medicine lies a patient, an individual. In recent decades, breakthroughs in omics technologies have empowered us to scrutinize the 100 trillion microbes, 36 trillion cells, 100 thousand metabolites, and 20 thousand genes and proteins that define an individual’s biological identity. Parallel advances in machine learning methods have enabled computational exploration of these incredibly large omics datasets. Yet, in our quest to transform omics technologies into precision health tools, a fundamental question remains: Which elements of our biological identities matter for our health? In other words, what constitutes a clinically relevant signal among biological noise? Where is the needle in an individual’s biological haystack?
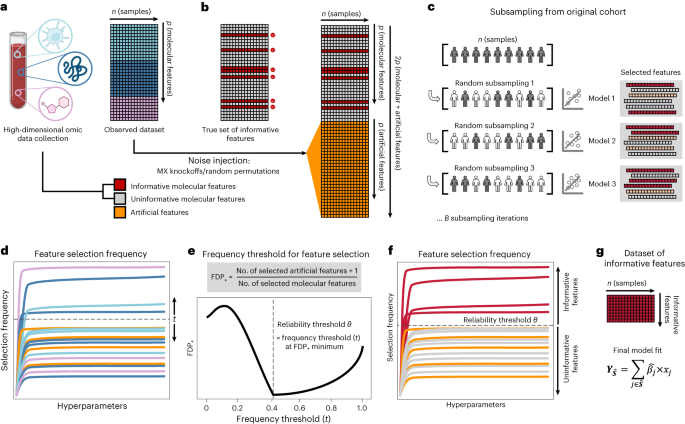
Our solution, a machine learning method called Stabl, bridges the domains of predictive modeling and artificial noise injection, introducing a data-driven threshold to separate the important bits from the noise. If our metaphorical haystack contains a few needles, perhaps ten or twenty, Stabl will highlight them—the consistent features that rise above an objective, data-driven threshold separating information from noise. These “stable” features are the ones that matter, the needles that can be harnessed to develop personalized medicine tools like scalable diagnostic tests and targeted therapies.
Big data…not so big sample size
It has become commonplace to wear smartwatches and biological sensors gathering continuous information from heartrate and temperature to blood sugar and sleep patterns. Similarly, clinical studies aiming to develop new diagnostics or therapies routinely include multiplex transcriptomic, metabolomic, or proteomic assays to gather biological information. So much accessible data sounds like a goldmine, yet the time and cost demands of omic data collection constrain the sample size. A typical study measures several thousands of omics features…in only dozens or, at most, hundreds of patients. Statistically, the numbers misalign and the “curse of dimensionality” looms.
Our machine learning framework builds on the sparsity-promoting regularization methods, such as Lasso, the Elastic Net, and their derivatives, to create multivariable predictive models while selecting a subset of features from high-dimensional omic datasets. Although these models introduce regularization parameters that control for the dimensionality to prevent overfitting, they are optimized for prediction rather than biomarker selection. Consequently, many features selected by these models turn out to be false discoveries, unsuitable for clinical translation. This is a major drawback, as clinical application requires identifying a manageable number of reliable (i.e., truly related to the clinical outcome) predictive biomarkers that can be measured using routine clinical assays.
The difficulty lies in discerning which elements of these models inform about clinical outcomes and which are merely background noise. Without this information, translating omics study results into actionable and scalable clinical tools remains challenging, leaving much of the promising “big data” untapped in storage. In essence, while high-content omics data holds enormous clinical potential, the central trial remains navigating this vast haystack to locate the needle.
Noise injection to answer a statistical challenge: uniting prediction with biomarker selection
To address the dual challenge of prediction and biomarker selection, we introduced simulated noise that mimics the inherent noise, or uninformative features, present in real-world datasets. This process of "noise injection" serves a crucial purpose: it allows us to calculate a data-driven, signal-to-noise “stability” threshold, below which the injected noise mingles with the inherent noise and above which the informative signal emerges.

Figure 1. Stabl Algorithm workflow. High-dimensional omic datasets often contain thousands of features, many of which do not relate to the outcome of interest. To identify the set of informative features, Stabl augments the original dataset with artificial features (using MX knockoffs or random permutations), under the assumption of exchangeability of the artificial features with the dataset’s uninformative features. Multivariable models are constructed using sample subsets in a bootstrapping fashion, and each feature’s selection frequency is calculated across bootstrap iterations. Calculating the minimum false discovery proportion using the artificial features allows determination of the best reliability threshold theta and identification of the set of informative features. The final Stabl model is built only on the subset of informative model features.
Noise injection coupled with our stability threshold provides a basis for determining which features are artifacts, or false-discoveries, and which are genuinely linked to the outcomes of interest. Building on the assumption that our simulated noise closely mirrors actual data noise, we have shown with a mathematical theoretical guarantee that our approach reduces the number of false discoveries. This means that the biomarkers we identify have a higher likelihood of being genuinely associated with the outcomes, making them reliable leads for clinical translation.
Why this matters: Identifying reliable and biologically interpretable biomarkers from real-world multi-omic datasets
To demonstrate Stabl’s ability to identify informative biological features, we tested it on synthetic datasets of various distribution, correlation, and outcome structures, where we had the advantage of knowing which features were informative. Across all conditions, Stable outperformed existing algorithms (including Lasso, Elastic Net, Adaptive Lasso, and Sparse-group Lasso) by selecting a set of biomarkers that overlapped more closely with the set of known informative features. We then set out to demonstrate whether Stabl improved the biomarker discovery process when tested on real-world clinical data, including five independent clinical studies spanning multiple clinical outcomes and omic technologies (transcriptomics, proteomics, metabolomics, microbiomics, and cytomics). In each clinical scenario, Stabl dramatically reduced the number of features selected compared to existing methods while maintaining a similar predictive performance.
By selecting fewer, more reliable biomarkers, Stabl greatly facilitated the biological interpretation of the predictive models and pointed at plausible targets for translation. For example, in a 30,000-feature cell-free RNA dataset collected during pregnancy to predict preeclampsia, Stabl achieved >83% predictive performance with over 20-fold reduction in the number of selected features compared to other predictive algorithms. In another study predicting when pregnant women would go into labor, Stabl distilled three omic datasets (metabolomic, proteomic, and single-cell mass cytometry), down to 27 predictive biomarkers. Finally, we tested our algorithm on a new multi-omic study predicting surgical site infections, one of the most devastating complications after surgery. Stabl honed in on a shortlist of proteomic and cytometric biomarkers predicting post-surgical infection that can be measured before surgery using clinically available assays.
Conclusion
The current heyday of extensive data collection has opened broad possibilities for personalizing medicine to enhance health outcomes. Stabl pushes the needle further by building upon advanced machine learning techniques, optimizing selection of a focused set of reliable biomarker candidates within predictive modeling. This advancement represents a crucial step in facilitating meaningful clinical translation. Beyond accelerating biomarker discovery within multi-omic research, Stabl’s modular framework is versatile and can be customized to various biological and clinical data types, including continuous biometric data and electronic health records, broadening its applications in healthcare and research.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in