Flipping the Script: How a Notorious Solid Tumor Suppressor Drives a High-Risk Leukemia Addiction
Published in Cancer, Cell & Molecular Biology, and Biomedical Research
For any research team diving into the genomics of pediatric leukemia, the Philadelphia chromosome–like (Ph-like) subtype of B-ALL represents one of the most frustrating walls in cancer biology. Even though we can map the genomic rearrangements driving its kinase signaling into overdrive, these cells consistently bypass targeted inhibitors. Looking at clinical data, where the three-year overall survival for these young patients has been stuck at a discouraging 65% plateau, is a stark reminder of what we're up against. It raises a fundamental question: if hitting the driving kinases directly keeps running into a wall of resistance, what deeper, hidden machinery is keeping these cells alive?
That exact frustration led our team to look past the usual oncogene suspects and at the machinery of chromatin remodeling, specifically the SWI/SNF complex.
In textbook solid tumors, the complex’s core catalytic engine, BRG1 (encoded by SMARCA4), acts as a protective tumor suppressor. It is frequently deleted or mutated because solid tumor cells want to get rid of it, prompting efforts to target its twin paralog, BRM (SMARCA2), via synthetic lethality.
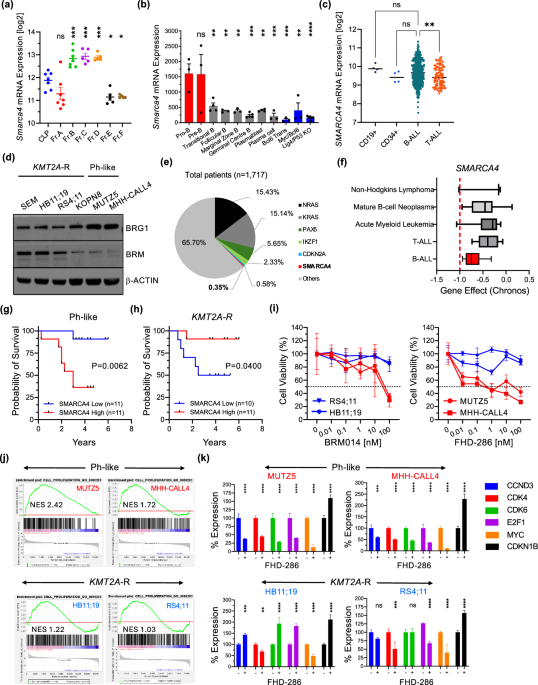
But when we mined clinical datasets, including the St. Jude Cloud cohort of 1,717 B-ALL patients, we hit a massive logical paradox. Across thousands of pediatric leukemic samples, SMARCA4 mutations were practically non-existent, appearing in just 0.35% of cases. In a classic tumor suppressor world, this makes zero sense. If losing a gene helps cancer grow, why would a leukemia cell guard its sequence with such flawless evolutionary conservation?
That was our "aha" moment. The paradigm was completely inverted: B-ALL blasts hadn't lost BRG1; they were profoundly addicted to it. They were actively hijacking wild-type BRG1 to stay alive, transforming our project into a rescue mission to unmask a context-dependent vulnerability.
Untangling the Lineage (and Dealing with Skepticism)
Proving a profound biological requirement is one thing, but proving its unique context is where the real work begins. When we pulled up CRISPR screens on the Cancer Dependency Map portal, the data confirmed that B-ALL models were deeply sensitive to SMARCA4 loss. However, our peers pushed back with fair skepticism: if every blood cancer needs this gene to survive, what makes your B-ALL story special?
To answer that, we looked at how a healthy B cell develops. We ran a meta-analysis of Smarca4 expression across normal mouse hematopoiesis, and the data instantly gave us our answer. Smarca4 expression spikes dramatically during early pro-B and pre-B cell development. Because B-ALL blasts are developmentally frozen at these exact progenitor stages, they inherit this natural, high-intensity requirement for BRG1, locking them into a state of developmentally hardwired dependency.
When we looked back at human data from the COG P9906 pediatric cohort, the clinical echo was impossible to miss. SMARCA4 expression was markedly elevated in B-ALL compared to healthy cells or T-ALL, clustering highest (after the TCF3-PBX1 group) within the high-risk Ph-like groups.
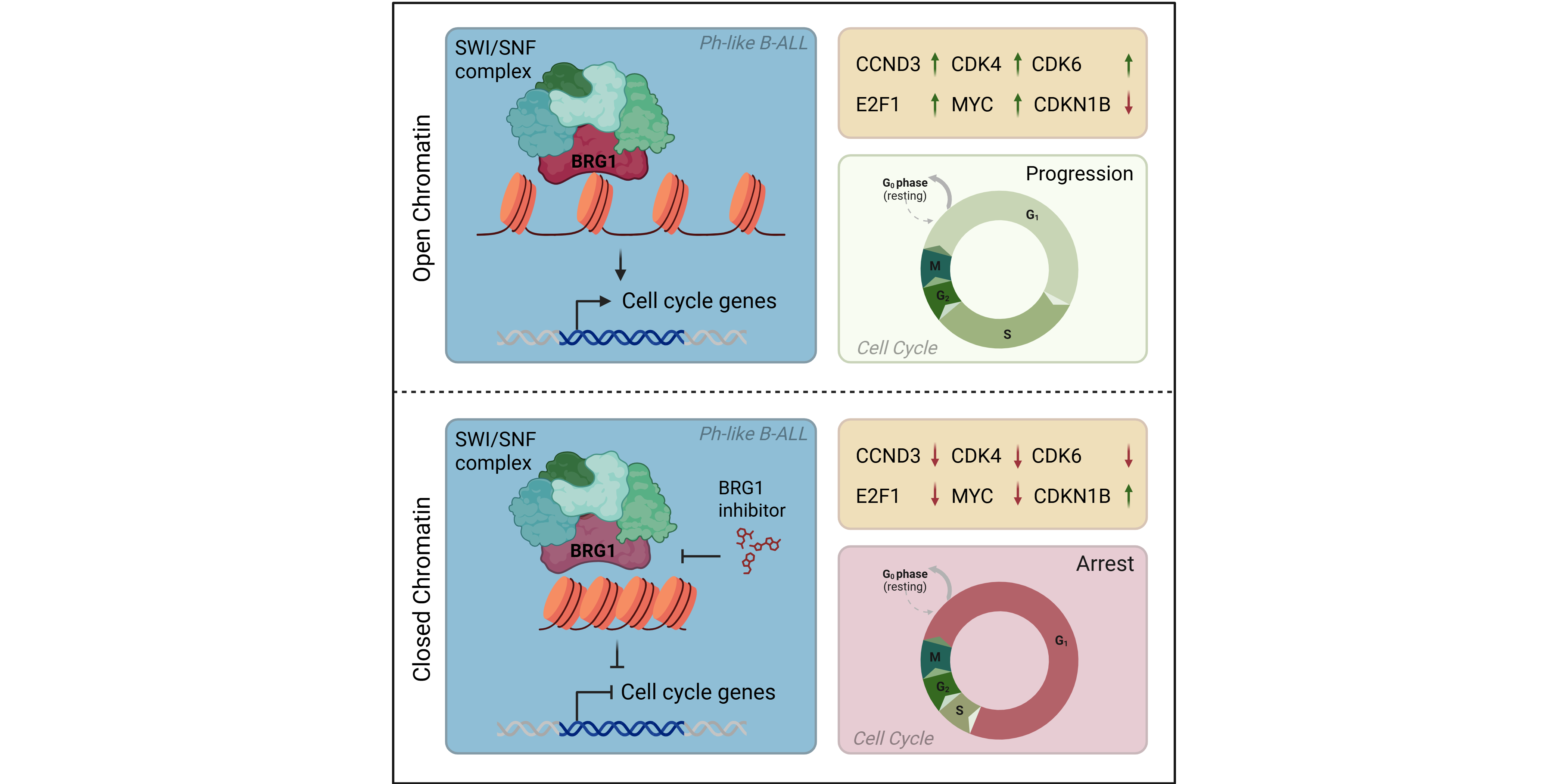
This subtype specificity lined up perfectly with patient survival. In the Ph-like group, high SMARCA4 expression tracked with significantly worse overall survival, but in KMT2A-rearranged (KMT2A-R) B-ALL, the trend completely flipped; higher expression actually meant better outcomes. This proved BRG1 isn't a generic housekeeping gene; its requirement is deeply wired into the specific, stage-dependent epigenetic landscape of the leukemic subtype.
Our lab validation bore this out beautifully. Western blots showed that Ph-like lines express a ton of BRG1 but have nearly zero BRM protein, while KMT2A-R lines express both. When we hit them with FHD-286, a potent small-molecule ATPase inhibitor, the Ph-like lines collapsed at single-digit nanomolar concentrations, while the KMT2A-R lines barely blinked. This was the highly context-dependent, stage-specific vulnerability we needed.
The Translational Horizon: Outsmarting Quiescence
Finding vulnerability is great, but the real test is figuring out how to use it. When we moved FHD-286 in vivo, oral monotherapy significantly extended survival and cleared out the leukemic burden in both cell-line and patient-derived xenograft (PDX) models.
But underneath the clean survival curves, the biology threw us a massive curveball. Our RNA-seq and flow cytometry revealed that blocking the SWI/SNF core doesn't actually kill the cells right away. Instead, it acts like a handbrake. By turning off critical cell cycle engines (CCND3, CDK4, CDK6) and turning up the brake protein p27 (CDKN1B), BRG1 inhibition forces the blasts into a deep, quiescent arrest. To make matters more complicated, our apoptosis arrays showed that these arrested blasts immediately throw up survival shields, upregulating anti-apoptotic proteins like Survivin and XIAP. In a clinic, this is dangerous; pausing the cells allows them to hide out in the protective bone marrow niche, wait out the treatment, and eventually drive a relapse.
We knew we had to force these sleeping blasts over the edge into actual cell death. So, we set up large in vitro synergy matrices, pairing FHD-286 with standard frontline chemotherapies. Using ZIP synergy modeling, we hit the jackpot when combining BRG1 inhibition with the corticosteroid dexamethasone or the spindle poison vincristine. The synergy scores were massive, with mean ZIP scores exceeding 10 and peaks hitting 20 to 31. On the flip side, combinations with doxorubicin or L-asparaginase were a total bust, showing narrow windows and flat-out antagonism.
The molecular logic behind the dexamethasone synergy makes total sense mechanically. Glucocorticoid receptors desperately need SWI/SNF complexes to physically open up condensed chromatin so they can turn on their gene programs. By using FHD-286 to disrupt BRG1-dependent chromatin structure, we're essentially lowering the defensive walls of the cell. We’re "epigenetically priming" the blasts, making even the most treatment-resistant Ph-like cells incredibly sensitive to a standard, cost-effective frontline therapy.
Ultimately, this gives us a concrete game plan for the clinic. We shouldn't use BRG1 inhibitors as a solo magic bullet. Instead, we can use them to pull the rug out from under leukemia's epigenetic defenses, transforming a stubborn state of cellular arrest into a wave of absolute apoptosis. We hope that this strategy will finally help us break through that frustrating 65% survival ceiling and give these kids a real, durable chance at a cure.
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in