How to Identify a Novel Class Kink-turn RNAs and their Mechanisms
Published in Bioengineering & Biotechnology

The eukaryotic genome is an RNA machine1, and discovering new class of non-coding RNAs (ncRNAs) with distinctive structural and sequence motifs and their regulatory mechanisms is vital to understanding how this RNA machine works. These ncRNAs often interact with RNA-binding proteins (RBPs) to carry out regulatory functions through distinct secondary or tertiary structures and sequence motifs. The kink-turn (K-turn) is the most prevalent three-dimensional (3D) RNA structural motif in messenger RNAs (mRNAs) and non-coding RNAs (ncRNAs)2-6. Moreover, the K-turn structure and its binding protein, 15.5K, play crucial roles in RNA metabolism and pathological processes2-6. However, the prevalence, mechanism, and function of K-turn structures in the transcriptome remain largely unknown due to the lack of effective experimental and computational methods for identifying RNAs with K-turn structures.
Inspiration for developing the RIP-PEN-seq and PEN-seq methods
Approximately 18 years ago, I developed the snoSeeker software to identify box C/D snoRNAs with forward K-turn motifs (fktRNAs) and H/ACA snoRNAs in the human genome7. Since then, I have been very curious whether there are new class of ncRNAs hidden within human genome. To satisfy my curiosity, I sought guidance from senior RNA experts by reading their personal reflections celebrating the 20th anniversary of the RNA journal in 20158. Additionally, I encouraged students to translate these reflections into Chinese and post them to a blog for all RNA researchers from China. Although all the reflections were very excellent, the most impactful one for me was written by Professor Joan A. Steitz9. Joan A. Steitz wrote "One is what I call the “black hole” of RNA biology: RNAs of 50 to 300 nucleotides (nt) have simply not been analyzed by deep sequencing."9. Inspired by this profound insight and I hypothesized that novel class of ncRNAs might hide within the “black hole”: RNAs of 20 to 1500 nt (but not limited to 50-300nt). To test this hypothesis, my lab spent about eight years to develop new deep sequencing methods and computational tools to explore the “black hole” of RNA biology and capture the full-length sequences and structual motifs of ncRNAs ranging from 20 to 500 nt (even up to 1500 nt).
Identification and definition of a novel class of ncRNAs with consensus backward K-turn and sequence motifs
For conventional RIP-seq and RNA-seq, the immunoprecipitated RNAs or long RNAs (especially for RNAs with length >= 50 nt) were often fragmented and then subjected to RNA sequencing library construction with random primer-based reverse transcription. As a result, the conventional RIP-seq and RNA-seq methods cannot identify the full-length of immunoprecipitated RNAs and long RNAs, and thus conventional RIP-seq and RNA-seq methods cannot discover the precise positions of motifs and consensus structural motifs at RNAs.
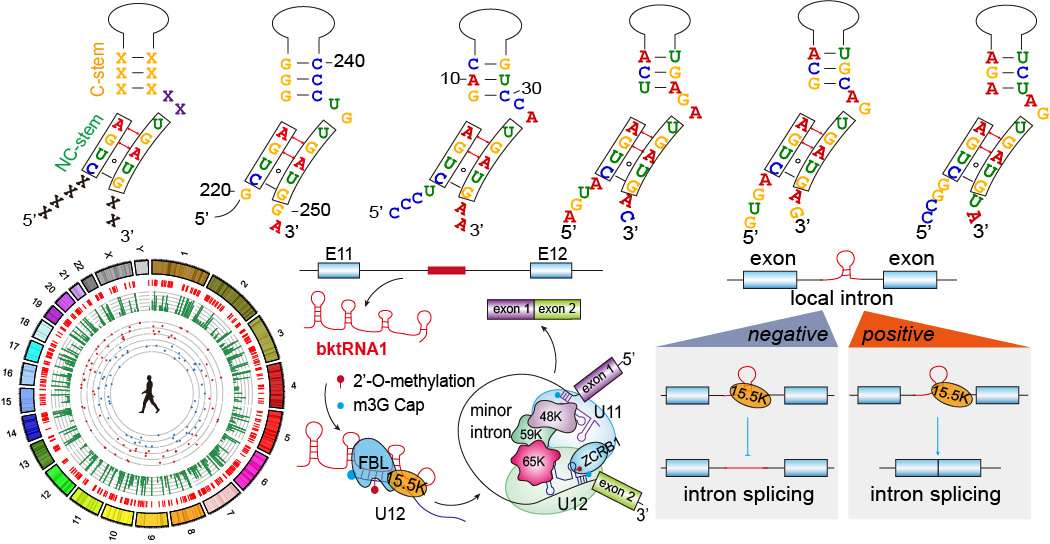
To overcome these limitations, we have developed an efficient RNA cloning method called RIP-PEN-seq/PEN-seq (Fig. 1) that uses dual RNA adapters and size selection and a series of advanced experimental strategies to capture both ends of any ncRNAs bound by the 15.5K protein. By implementing this new technique and a new computational algorithm (kturnSeeker), we discovered a significant number of backward ktRNAs, as well as more than 600 novel forward ktRNAs (fktRNAs). Through structural and sequence motif enrichment analysis of these new backward ktRNAs, we found that most of them have the following unique characteristics: (1) they have a backward K-turn structural motif; (2) the K-turn structural motifs of the majority of bktRNAs are located at 4 nt and 2 nt from the RNA 5’ end and 3’ end, respectively; (3) The 5' end of the backward K-turn structural motifs contains CUGA motif and the 3' end contains UGAUG motif. Using RIP-PEN-seq and kturnSeeker to mouse cells, we also found that bktRNAs in mice shared these characteristics with human bktRNAs. In addition, we validated these unique characteristics through the analysis of SHAPE reactivity signals generated by our 15.5K RIP-PEN-SHAPE-MaP methods. Therefore, based on these unique characteristics, we have named them as a novel class of ncRNAs with consensus backward K-turn structural motifs (bktRNAs).
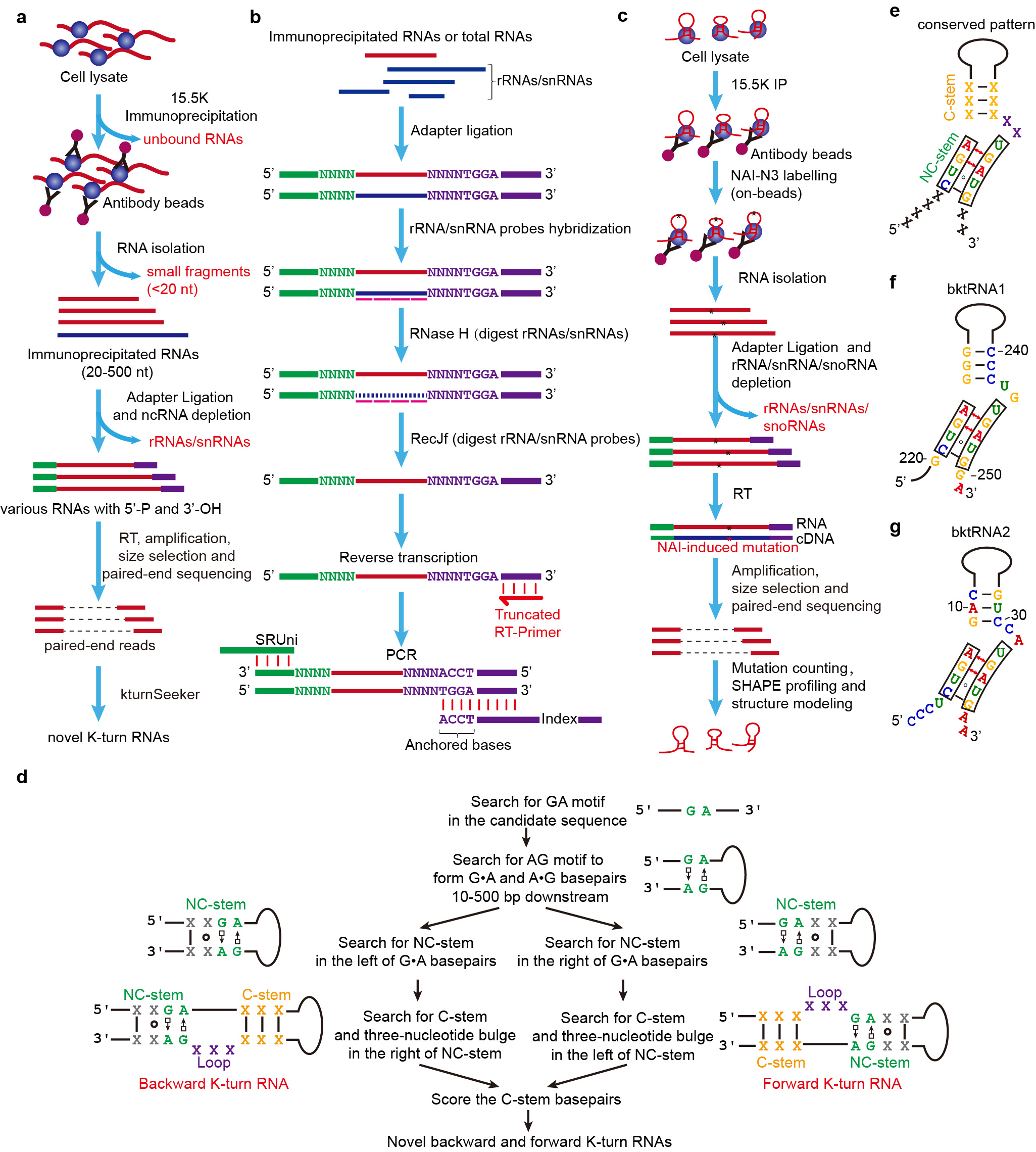
Fig. 1 | New experimental and computational methods for identifying kink-turn RNAs. a, Procedure for the construction of RIP-PEN-seq libraries. b, Diagram of RNase H-based rRNAs/snRNAs depletion for the construction of RIP-PEN-seq or PEN-seq library. c, Workflow for the construction of 15.5K RIP-PEN-SHAPE-MaP libraries. d, KturnSeeker core algorithm workflow. KturnSeeker was developed to identify and quantify forward (fktRNAs) and backward ktRNAs (bktRNAs) from RIP-PEN-seq data. e, Gene model of bktRNA identified from RIP-PEN-seq data. f, The predicted secondary structure of the bktRNA1. g, The predicted secondary structure of the bktRNA2.
Function and mechanism of the highly conserved and chimeric bktRNA1
ncRNAs predominantly exert their functions through forming complementary base pairs with specific RNA molecules9. Based on this concept of RNA–RNA base-pairing9, we developed starBase platform10, 11, which has been cited more than 15000 times in Google Scholar. This platform integrates almost all public CLIP, CLASH and PARIS sequencing data to decode the RNA-RNA interactome10, 11. Using tools developed in our starBase, we analyzed chimeric reads in the 15.5K and FBL CLASH and PARIS sequencing data and discovered that U12 snRNA is the direct target of bktRNA1. We also found that BktRNA1 is indispensable for 2’-O-methylation of A8 of U12 snRNA by using our RMBase platform12, infrared primer extension (irPE), and gain and loss of function experiments. Depletion of bktRNA1 affected more than 75% of U12-type introns; at least 37% of the retained introns had significant changes (P value <0.05), indicating bktRNA1 and Am8 in U12 snRNA are crucial for the fidelity of U12-type splicing in human cells. We further confirmed the 2’-O-methylation at A8 in U12 snRNA guided by bktRNA1 is critical for the recruitment of ZCRB1 to the U11-U12 di-snRNP complex as well as for the splicing of U12-type introns by using Northern Blot, ChIRP,RNA pull-down and RNA EMSA experiments (Fig. 2).
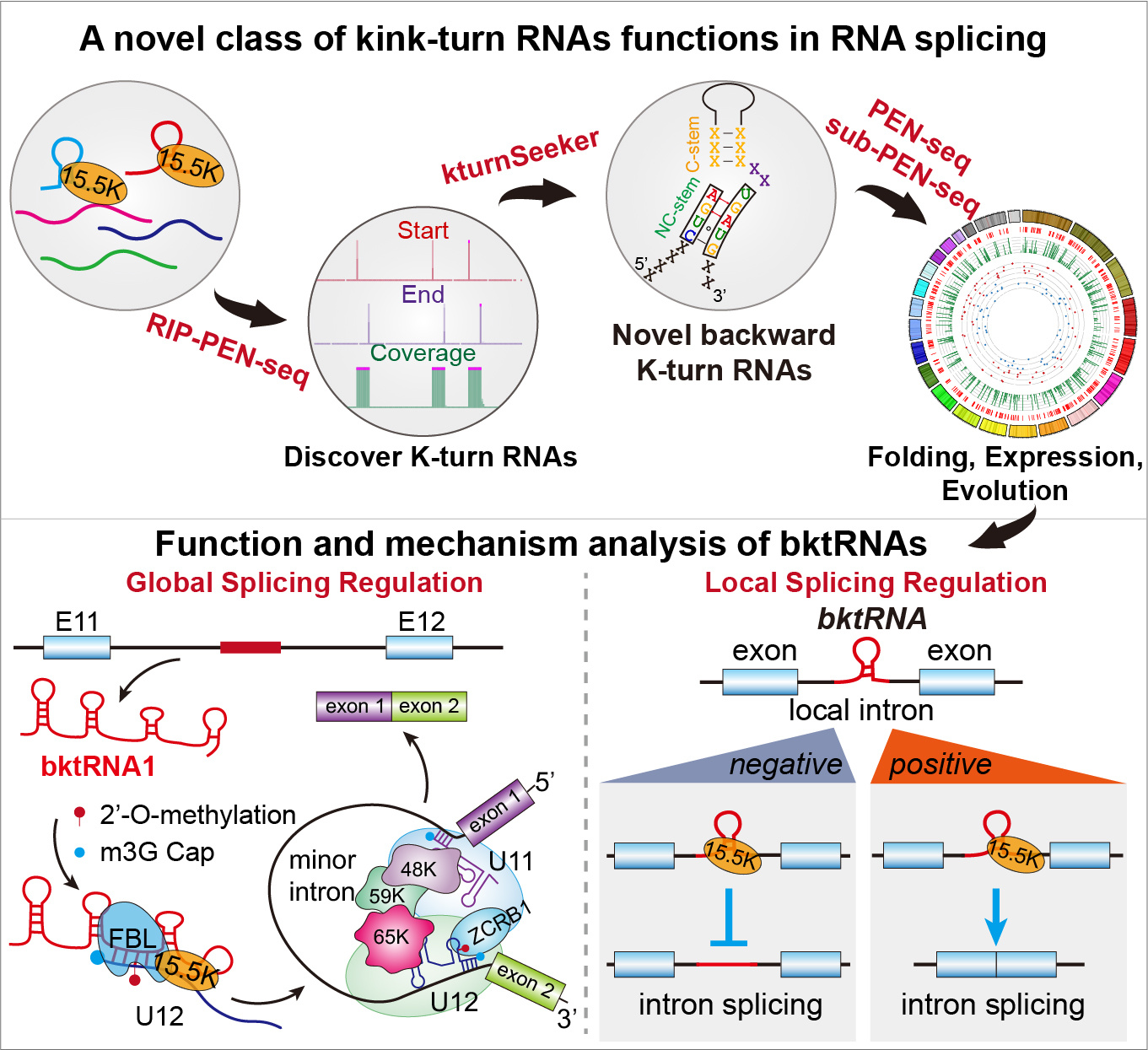
Fig. 2 | The workflow illuminating how to identify a novel class of ncRNAs with consensus backward K-turn structural motifs (upper panel). Proposed model showing the functions and mechanisms of bktRNAs (bottom panel).
The potential function of the consensus backward K-turn structural motifs
We have demonstrated that the consensus backward K-turn structural motifs are indispensable for the local regulation of intron splicing by bktRNAs through using prime editing technology and GFP reporters (Fig. 2). We also found that these backward K-turn structural motifs of bktRNAs are indispensable for avoiding degradation by exonucleases, the processing and maturation of bktRNAs. Unlike box C/D fktRNAs (fktRNAs), the CUGA motif is at the 5' end of backward K-turn motifs, so the backward K-turn motifs may not be used as a marker for selecting functional regions, but rather serve as a structural element for stabilizing RNA. Interestingly, we identified a consensus backward k-turn structural motif in an ncRNA previously discovered by Joan A. Steitz's lab, which explains the long-standing suspicion why its 5' end is not degraded by exonucleases (unpublished data). These findings strongly support the significance of the consensus backward K-turn structural motif for the stabilization, biogenesis and function of bktRNAs.
What's next?
Our study has raised many intriguing scientific questions pertaining to this novel class of bktRNAs that require further elucidation. For example, what are the underlying mechanisms governing the positioning of the backward K-turn structural motifs at 4 nt and 2 nt from the RNA 5' and 3' termini, respectively? Do other bktRNAs function in guiding 2'-O-Me modification or other types of modifications, such as U13 box C/D fktRNA mediating ac4C modification? Additionally, what factors contribute to the selection of 5' CUGA and 3'-UGAUG motifs as the central components of the consensus backward K-turn structure? Whether there are different structural basis and molecular mechanism for the interaction between bktRNA and fktRNA and 15.5K protein? Apart from the binding protein 15.5K, what other proteins may interact with the backward K-turn structural motif? Is the unique backward K-turn structure exclusive to vertebrates or do other organisms, such as insects, nematodes, plants and archaea, also possess similar characteristics? Whether the 2'-O-Methylation on U12 snRNA mediated by bktRNA1 also exists in other organisms, such as insects, nematodes, plants and archaea? What are the biological functions of the more than 650 novel fktRNAs we identified? Does 2'-O-Methylation on the snRNAs of major splicesome mediated by ktRNAs also affect the splicing of U2-type introns? Are there any new classes of ncRNAs hidden in humans and other organisms?
Revisiting the Study of ncRNAs
Our study provides some innovative concepts for the study of ncRNAs as follows: 1. The focus of ncRNA research should not be confined to small RNAs (e.g. miRNAs and piRNAs) with length less than 50nt, but rather include various types of structural ncRNAs with a broad range of lengths (20-1500nt). 2. Instead of solely examining the expression of ncRNAs through sequencing, we propose to simultaneously investigate both the expression level and precise full-length sequence of ncRNAs. 3. In addition to studying the primary sequence of ncRNAs, it is essential to explore the sequence motifs, the secondary and tertiary structural motifs, in order to fully understand their functions and mechanisms. Finally, These innovative concepts and our methods (RIP-PEN-seq, PEN-seq and RIP-PEN-SHAPE-MaP) can be combined to discover novel class of ncRNAs in diverse organisms and explore the roles and underlying mechanisms of ncRNAs , thereby enriching our understanding of RNA molecules.
In summary, our findings characterize a novel class of small RNAs and uncover another layer of gene expression regulation that involves crosstalk among bktRNAs, RNA splicing and RNA methylation. We anticipate that this study will provide new avenues for discovering other bktRNAs or ncRNAs and unraveling their functions and mechanisms in cells and diseases.
References
- Amaral, P.P., Dinger, M.E., Mercer, T.R. & Mattick, J.S. The eukaryotic genome as an RNA machine. Science 319, 1787-1789 (2008).
- Klein, D.J., Schmeing, T.M., Moore, P.B. & Steitz, T.A. The kink-turn: a new RNA secondary structure motif. EMBO J 20, 4214-4221 (2001).
- Lilley, D.M. The K-turn motif in riboswitches and other RNA species. Biochim Biophys Acta 1839, 995-1004 (2014).
- Schroeder, K.T., McPhee, S.A., Ouellet, J. & Lilley, D.M. A structural database for k-turn motifs in RNA. RNA 16, 1463-1468 (2010).
- Edery, P. et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 332, 240-243 (2011).
- He, H. et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 332, 238-240 (2011).
- Yang, J.H. et al. snoSeeker: an advanced computational package for screening of guide and orphan snoRNA genes in the human genome. Nucleic Acids Res 34, 5112-5123 (2006).
- Nilsen, T.W. Twenty years of RNA: then and now. RNA 21, 471-473 (2015).
- Steitz, J. RNA-RNA base-pairing: theme and variations. RNA 21, 476-477 (2015).
- Yang, J.H. et al. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 39, D202-209 (2011).
- Li, J.H., Liu, S., Zhou, H., Qu, L.H. & Yang, J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42, D92-97 (2014).
- Xuan, J.J. et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res 46, D327-D334 (2018).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in