Integrating Single-cell Atlases with scDREAMER
Published in Protocols & Methods and Genetics & Genomics
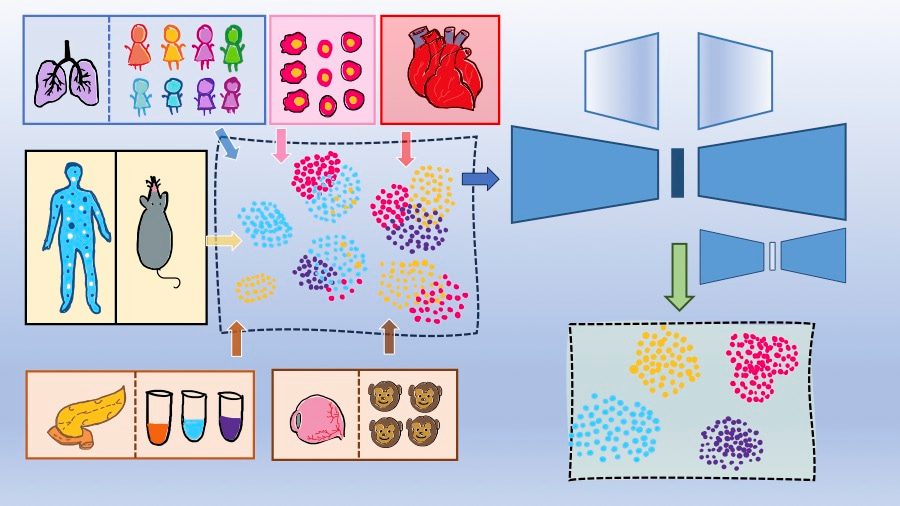
Data integration: an essential prerequisite of Single-Cell RNA-seq analysis
Recent advances in single-cell sequencing techniques coupled with large-scale collaborative initiatives such as the Human Cell Atlas [1] have transformed the discovery and comprehension of various cell types and their distinct functional states. The steady growth of single-cell experiments have resulted in the generation of complex datasets that can include samples contributed by different laboratories, generated across tissue locations, time and conditions [2, 3]. Integration of such heterogeneous datasets can provide comprehensive insights into the cellular states and expression programs that cannot be obtained from individual datasets [4, 5]. However, datasets collected from different sources harbor complex, nested batch effects. An efficient data integration method needs to overcome such batch effects while preserving genuine biological variations between batches. While integration methods have been developed that rely on mutual nearest neighbors (MNNs) [6] or deep learning models [7] for integrating multiple batches, the existing approaches face challenges due to low-quality MNNs, large memory consumption and sub-optimal integration as obtained using traditional autoencoder models. A comprehensive benchmarking study [8] of the existing integration methods highlighted the need for developing new integration methods which can perform well in correcting batch effects and preserving biological variations. To address these, we therefore aimed to develop a new efficient data integration method.
The proposed method scDREAMER
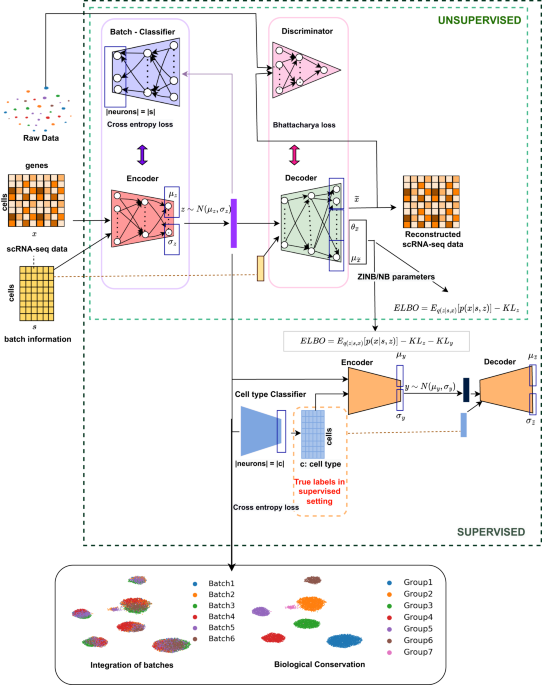
We developed scDREAMER (single cell Deep geneRative intEgrAtion Model with advErsarial classifieR), a data-integration framework that employs deep generative models and adversarial training for both unsupervised and supervised (scDREAMER-Sup) integration of multi-batch single-cell data (Figure 1). The unsupervised version of scDREAMER utilizes an adversarial variational autoencoder, a type of deep generative model for learning the low-dimensional cellular embeddings. The batch effects are removed from these low-dimensional representations with the help of a batch classifier neural network which is trained adversarially with the variational autoencoder. The supervised version, scDREAMER-Sup, employs an additional variational autoencoder and a cell-type classifier neural network to utilize available cell-type annotations for a semi-supervised or supervised inference of low-dimensional cellular representations.

Evaluation and Biological Applications
We applied scDREAMER to a diverse set of integration tasks on datasets consisting of up to 1 million cells and 147 batches (Figure 2). These complex data integration tasks were designed to evaluate scDREAMER’s ability to handle a variety of integration challenges such as the presence of skewed cell types among batches (pancreas integration), nested batch effects (lung and human immune integration), large number of batches (healthy human heart integration with ~0.5 million cells) and atlas level integration (human and mouse integration).
We compared scDREAMER's performance in batch-correction and conservation of biological variation against that of 11 state-of-the-art unsupervised and supervised integration methods including scVI, Harmony, Scanorama, and scANVI which were the best performers according to the most recent benchmarking. For performance comparison, we used different metrics for assessing bio-conservation and batch-correction as well as composite score metrics that assess the holistic performance of a method.
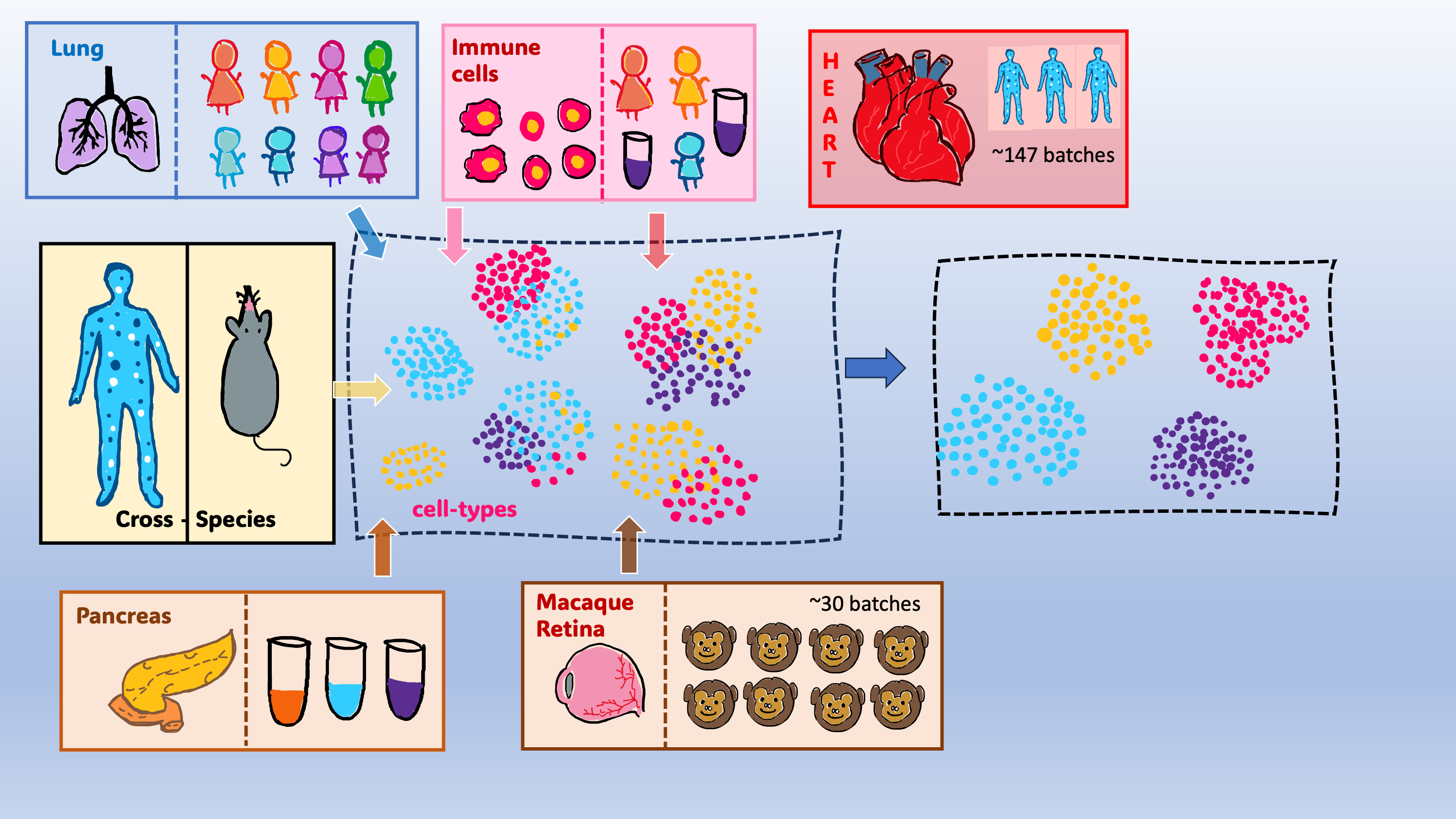
We observed that both scDREAMER and scDREAMER-Sup outperformed 11 state-of-the-art unsupervised and supervised integration methods respectively in batch-correction and conservation of biological variation across different integration tasks. scDREAMER also demonstrated high accuracy for the integration of a large number of batches and atlases from different species despite the small number of shared cell types. scDREAMER-Sup also performed superior to all other methods in predicting the cell type labels for the cells missing annotations. Particularly for the tasks involving a large number of cells and batches, scDREAMER outperformed the other methods by a large margin, which highlights scDREAMER’s scalability to a large number of batches and millions of cells. We also delved deeper into the biological insights obtained by scDREAMER from the human immune integration task where it identified subtypes of dendritic cells which the other methods failed to capture. We also observed that running scDREAMER was faster compared to certain other deep learning methods. This coupled with higher accuracy of scDREAMER particularly makes it an important data integration method as the deep learning-based methods enable the inference of latent cellular embeddings as well as corrected expression profiles which are required for several downstream applications such as trajectory inference or differential expression analysis. As more cell atlases are generated from different species, we believe that scDREAMER will be a suitable tool for the biologists for performing integration of cross-species datasets for the discovery of cell types.
Future Directions
An important future direction would be to explore the unsupervised treatment of batch information and whether the hierarchical structure between different batch information can be utilized when it exists (e.g. when cells from a single donor but multiple organs are present in the atlas). While we restricted our analysis to the integration of scRNA-seq datasets, our deep generative model encompasses a general framework which can accommodate other omics datasets and we plan to extend the framework of scDREAMER to multiomic datasets. Another future direction will be to extend the deep generative model to also learn gene embeddings for better interpretability. Finally, given the rapid generation of atlas-level single-cell datasets across multiple organs and species, we anticipate that applications of supervised and unsupervised models of scDREAMER will enable accurate integration of single-cell atlases for the exploration of different biological systems.
References
[1] Rozenblatt-Rosen, O., Stubbington, M. J., Regev, A. & Teichmann, S. A. The human cell atlas: from vision to reality. Nature 550, 451–453 (2017)
[2] Consortium, T. M. et al. A single cell transcriptomic atlas characterizes aging tissues in the mouse. Nature 583, 590 (2020)
[3] Gehring, J., Hwee Park, J., Chen, S., Thomson, M. & Pachter, L. Highly multiplexed single- cell RNA-seq by DNA oligonucleotide tagging of cellular proteins. Nature Biotechnology 38, 35–38 (2020)
[4] Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology 15, e8746 (2019).
[5] Pandey, K. & Zafar, H. Inference of cell state transitions and cell fate plasticity from single-cell with MARGARET. Nucleic Acids Research 50, e86–e86 (2022). URL https://doi.org/10 .1093/nar/gkac412
[6] Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcrip- tomes using Scanorama. Nature Biotechnology 37, 685–691 (2019).
[7] Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nature Methods 15, 1053–1058 (2018)
[8] Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nature Methods 1–10 (2021)
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in