Investigating subcellular transcriptional programs in triple negative breast cancer cells
Published in Cancer, Genetics & Genomics, and Mathematics
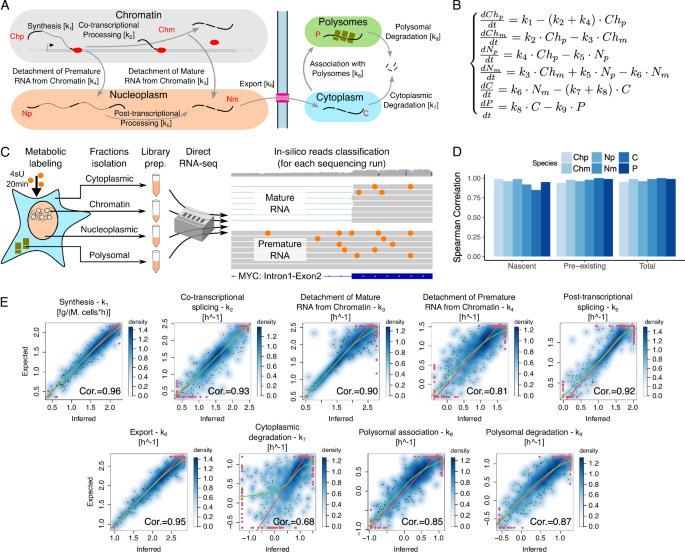
Gene expression is the process by which information stored in the DNA is transformed into functional products, called proteins, which perform most of the work in cells. The process of gene expression goes through two major steps: transcription, in which DNA is recoded in another alphabet to become RNA, and translation, in which instructions encoded in RNA are used to make proteins. It is commonly thought that RNA abundance is a good proxy for transcription efficiency. However, the RNA in the nucleus reaches the cytoplasm after a long journey full of stages (RNA dynamics), each of which influence the net amount of RNA that will be translated into proteins. Until recent times, the state of the art in terms of RNA dynamics was limited to the study of RNA synthesis, processing and degradation. When I arrived in Pelizzola’s lab, at the Centre for Genomic Sciences in Milan, to start my PhD in computational biology, I was captivated by the project of Prof. Pelizzola and Dr. Mattia Furlan to deepen the characterization of RNA life cycle by trying to estimate the rates of processing (k2-k5), export (k6), polysomal association (k8) and decays (k7,k9) (see scheme of the RNA life cycle in Figure 1) and I immediately started working on it.
To be able to quantify RNA dynamics, we used a mixed approach , that we called Nanodynamo, combining mathematical modeling and an experimental set-up providing the data to fit the model. More in detail, the mathematical model is able to estimate the kinetic rates (i.e. the efficiency of transcription, export, etc.) starting from the abundance of newly-synthetized, premature, and total mRNA within chromatin, nucleoplasm, cytoplasm and polysomes. The experimental set up relies on cellular fractionation and Nanopore single-molecule RNA sequencing.
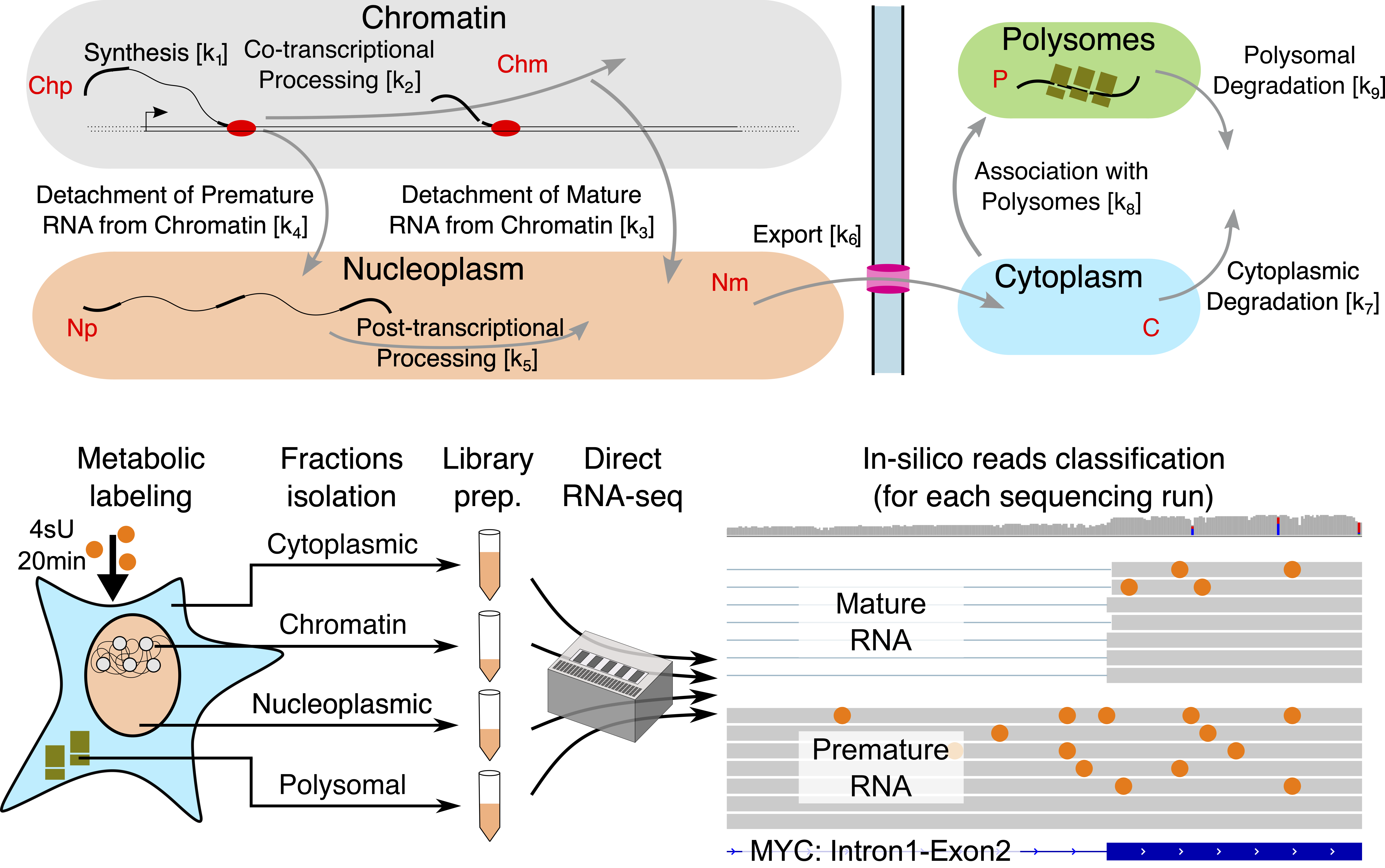
Figure1: Schemes of the RNA life cycle (up) and of the experimental set-up (down).
The hybrid nature of this work is what I loved the most: since I have a quantitative background (MSc in Physics of Complex Systems), my main task was the development of the computational part. However, I was so willing to not miss details in any part of the project that I started to do some experiments under the patient guide of Lucia Coscujuela Tarrero, the other first author of this work. Going back to science, to begin with we conducted our experiments in untreated triple negative breast cancer cells, which are known to be altered in transcriptional programs, and surprisingly we found out that most of the nuclear steps of RNA life cycle showed a bimodal distribution in terms of kinetic rates. At first, we thought that these bimodal distributions could represent an artifact but it turned out they were an intrinsic feature of the data. Once estimated the kinetic rates in untreated cells, we wanted to see if we could capture changes in the dynamics by perturbing RNA life cycle. To do so, we impaired splicing, export and translation and discovered not only that the model was able to capture these changes but also that perturbing one step of RNA life cycle has broad effects on all the other steps.
After collecting kinetic rates from both untreated and perturbed cells, we observed that some rate pairs consistently varied in synchrony, either in the same or in the opposite direction. This led us to hypothesize that coupling mechanisms might influence different stages of the RNA life cycle. To investigate this hypothesis, we performed a correlative analysis and built a network of couplings for each treatment. We found out that most of the couplings were shared among treatments and we sought to identify regulatory factors potentially responsible for each of the detected couplings. Doing so, we discovered that there were some common RNA binding proteins and transcription factors mediating couplings among treatments.
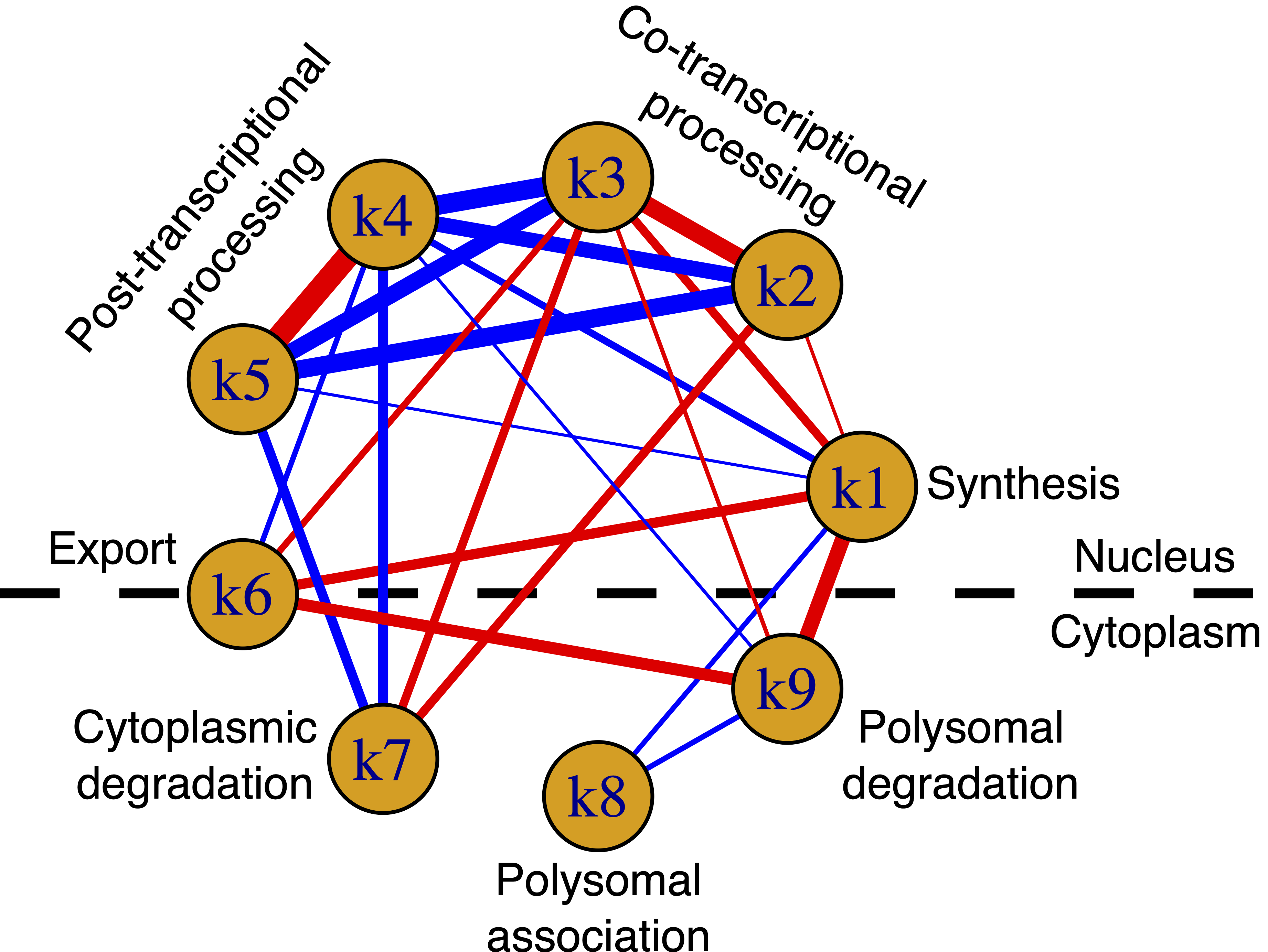
In conclusion, we developed a tool that provides unprecedented insight into sub-cellular RNA dynamics and coupling interactions between stages of the RNA life cycle. As a first application of Nanodynamo, we used it to characterize at unprecedented resolution the gene-expression programs of triple-negative breast cancer cells and their rewiring following drug treatments. Our work offers a foundation for future studies on specific coupling mechanisms, system-level properties, and vulnerabilities in RNA metabolism, particularly in disease contexts.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in