Looking for What Isn’t There: Exploring Gene Loss in Squamate Lineages
Published in Genetics & Genomics

In myth, lizards and snakes decide immortality or victory. For instance, snakes appear in the ouroboros of ancient Egypt, a serpent consuming its own tail, as well as in the Nāga traditions of India. Similarly, from Amazonian traditions where the lizard is regarded as the king of beasts, to Indian folklore where a monitor lizard becomes an agent of victory, squamates recur as powerful cultural symbols. Yet evolution has written an interesting, more reductive story in their genomes.
 Evolutionary Gene Losses
Evolutionary Gene Losses
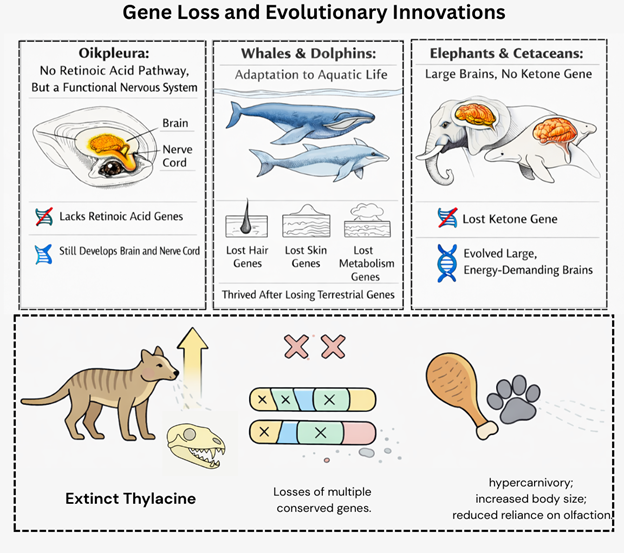
For a long time in the past, it was often assumed that losing genes produces streamlined or simplified biological systems. However, comparative genomics tells a different story. For example, a small marine animal called Oikopleura dioica can form a brain and nerve cord even though it lacks the genes for retinoic acid signalling, a pathway long thought necessary for nervous system development. Whales and dolphins lost many genes used by land mammals as they adapted to life in water, including genes involved in skin, hair, and metabolism. Even elephants and cetaceans evolved large, energy-demanding brains after losing a gene required for ketone production, a process once assumed to be essential for brain function during fasting. Not just extant species, but the extinct thylacine experienced ancestral losses of multiple conserved genes, coinciding with shifts toward hypercarnivory, increased body size, and reduced reliance on olfaction [1]. Together, these cases show that evolution does not always move forward by adding genes but often by discarding them and reorganising existing biological systems.
Most studies of gene loss have focused on individual genes or specific functional systems, leaving open the question of how widespread and structured gene loss is across entire genomes. Squamata, the reptilian order encompassing lizards, snakes, and amphisbaenians, represents one of the most diverse vertebrate groups with over ~11,000 described species, second only to ray-finned fishes in species richness. Originating over 242 million years ago, this group has since radiated into nearly every terrestrial habitat, from deserts to rainforests [2]. This far-reaching ecological success is mirrored by an exceptional diversity of form and function, including repeated limb loss, venom evolution, immune specialisation and reproductive innovation. Despite this diversity and their unique biological traits they have remained comparatively underexplored in the context of gene loss via segmental deletion. Hence, we set out to map the global pattern of gene loss across squamates and identify the structural mechanisms driving these losses.
Technological and methodological advancements in the field of gene losses due to segmental deletions
Comparative genomics identifies many candidate gene losses, but limited experimental throughput and difficulty in faithfully recapitulating natural deletions constrain causal inference. Hence, progress of most gene loss studies ultimately depends on accurate annotations, scalable pipelines, and standardized, computable phenotypic data, without which ancestral gene losses remain hard to detect and interpret.
Advances in long-read sequencing and the availability of chromosome-level assemblies—driven by large-scale international consortia such as the Vertebrate Genomes Project (VGP) [3] and the Earth BioGenome Project (EBP), alongside numerous species-specific initiatives—have largely helped to eliminate artifacts that previously mimicked gene loss, mainly due to high GC-content. At the same time, annotation-independent orthology and alignment-based frameworks such as TOGA [4] now allow systematic identification of gene losses due to frame-disrupting changes, even in genomes lacking curated annotations. Together with phylogeny-aware divergence models that incorporate insertions and deletions, these developments have redefined gene loss from a by-product of annotation gaps into a directly detectable and testable evolutionary signal.
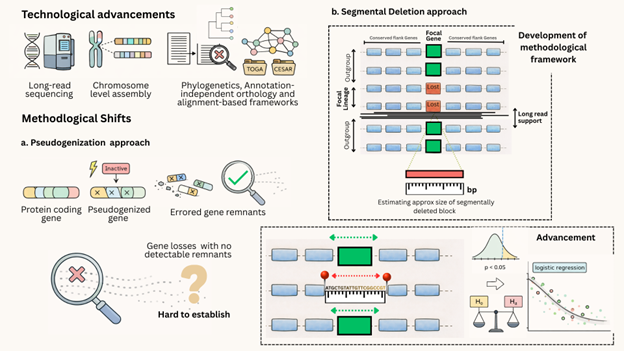 Early studies of gene loss focused on pseudogenization [5], where relaxation of selective pressure allows deleterious mutations to accumulate until a gene becomes nonfunctional. Subsequent deletion-biased genome evolution progressively erodes pseudogene remnants, often leaving no detectable sequence. This gradual erosion makes ancient gene losses difficult to identify using annotation- or mutation-based approaches alone. Hence, over short evolutionary timescales—such as between closely related species—gene losses can often be traced through recognizable pseudogenes. However, order wide gene losses i.e across deeper timescales (>200 million years in case of Squamata), persistent deletion bias means that most ancient losses are expected to have been fully excised from genomes, leaving no molecular trace and requiring alternative comparative frameworks for detection.
Early studies of gene loss focused on pseudogenization [5], where relaxation of selective pressure allows deleterious mutations to accumulate until a gene becomes nonfunctional. Subsequent deletion-biased genome evolution progressively erodes pseudogene remnants, often leaving no detectable sequence. This gradual erosion makes ancient gene losses difficult to identify using annotation- or mutation-based approaches alone. Hence, over short evolutionary timescales—such as between closely related species—gene losses can often be traced through recognizable pseudogenes. However, order wide gene losses i.e across deeper timescales (>200 million years in case of Squamata), persistent deletion bias means that most ancient losses are expected to have been fully excised from genomes, leaving no molecular trace and requiring alternative comparative frameworks for detection.
These limitations motivated a shift toward structural explanations for gene loss, particularly mechanisms that remove entire loci rather than leaving detectable pseudogenic remnants.
The initial methodological framework for studying gene loss via segmental deletion originated with locus-specific studies, such as the concurrent loss of interacting ciliary genes CFAP46 and WDR93 in galliform birds, which demonstrated a complete deletion and the fragmented remnants of a partner gene [6]. This initial paradigm—combining homology searches, conserved microsynteny, and long-read assembly validation—has since broadened with observations of multiple mechanistic routes, such as promoter deletion initiating coding-sequence erosion (CXCR6 in songbirds) [7] or the loss of one partner leaving a compensatory footprint on its receptor (IL34/CSF1R). Together, these cases confirm segmental deletion as a recurring and plausible mechanism of gene loss, challenging earlier interpretations that missing genes merely reflect annotation gaps or localization to genomic "dark matter."
Building on this foundational framework and moving beyond the mere detection of segmental deletions, the present study introduces a statistical framework to formally test this mechanism of gene loss. We employed phylogenetic logistic regression to model the probability of gene loss as a function of normalized intergenic distance, while explicitly controlling for shared evolutionary history. This approach transforms synteny disruption from a descriptive observation into a quantifiable, lineage-aware signature of segmental deletion.
Our Approach for the present study
We began by systematically comparing 261 squamate genomes against other vertebrates to identify genes consistently present in outgroups but absent from all annotated squamates. This yielded 53 candidate "unretrieved orthologs"—putative squamate-specific losses.
Simply not finding a gene in an assembly is not proof of loss; it could be missing due to poor assembly quality, high sequence divergence, or genomic relocation. To rule these out, we deployed a battery of validation checks.
We analysed the chromosomal neighbourhoods of each candidate gene by comparing syntenic blocks in squamates to those in other vertebrates. This allowed us to distinguish between conserved and disrupted synteny, classifying losses as resulting from segmental deletions, interchromosomal rearrangements, or intrachromosomal rearrangements. Further statistical validation came from analysing intergenic distances and performing phylogenetic logistic regression, which confirmed signatures of segmental deletion.
We then conducted sensitive sequence searches across assembled genomes, raw short- and long-read sequencing datasets from squamates, used HybPiper to assemble candidate gene sequences directly from raw sequencing reads, and additionally screened transcriptomic data from multiple tissues to test for residual or transcriptionally active sequences. Wherever similar sequences were detected, phylogenetic analyses were used to distinguish ortholog loss without retained paralogs from cases of ortholog loss with paralog retention. For conserved synteny loci, we verified genome assembly integrity using long-read data to ensure that absences were not caused by local assembly errors. In parallel, we examined GC content and sequence divergence for unretrieved orthologs across vertebrate species available in NCBI to ensure that losses were not biased by technical factors such as high GC-rich “dark” genomic regions.
What we found ?
This rigorous, multi-evidence framework allowed us to move from a list of unretrieved orthologs to a confident portrait of programmed genomic reduction. Most of the unretrieved orthologs are found to be GC-neutral, and we could classify the 53 losses into three groups: 14 genes lost entirely without a paralogous backup; 15 genes lost with a retained paralog likely providing compensation; and 24 genes that remain unretrieved orthologs, representing highly diverged or ambiguous cases.
Our analyses further revealed that gene loss in squamates is driven by two closely related structural mechanisms: segmental deletions, where entire genomic intervals are physically excised, and chromosomal rearrangements, which break conserved synteny and relocate genes into unstable genomic contexts, likely leading to their structural and functional erosion. Based on these integrated analyses, we categorised the 53 lost genes into two mechanistic groups: 36 lost via segmental deletion and 17 lost via chromosomal rearrangement.
Importantly, Unlike birds, where missing genes are often confined to GC-rich microchromosomes, squamate gene losses are mechanistically diverse. Many occur in conserved syntenic blocks in non-squamate vertebrates and map to regions that have undergone extensive intra- or interchromosomal rearrangements in squamates.
Of the 53 investigated unretrieved orthologs, four are found to be involved in macrophage activation and polarisation—IL34, STAP1, LAPTM5, and TNIP2—while others play key roles in immunity, development, including pathways implicated in tail development and regeneration metabolism, cellular processes, and neuronal and musculoskeletal functions.
Why does it matter ?
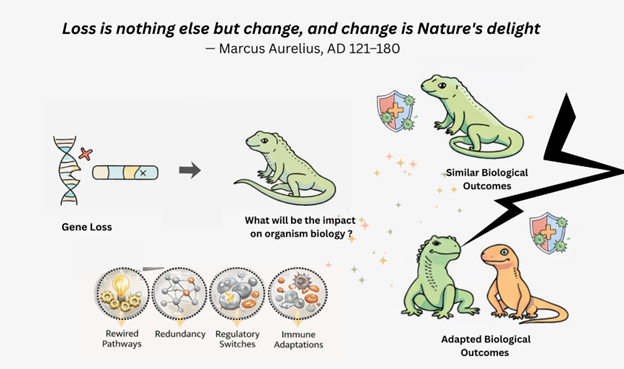
Without the comparative signal provided by gene loss, many of these lineages would not appear exceptional at all. It is only when a conserved gene is lost that a lineage—squamates, in this case—stands out, compelling us to understand what alternative evolutionary solutions have arisen
This shift in perspective moves the focus beyond cataloguing missing genes to understanding how squamates function normally despite the absence, modification, or erosion of genes that are conserved and often functionally important in other vertebrates. For instance, the loss of immune-related genes such as IL34, STAP1, LAPTM5, and TNIP2 in squamates raises the possibility that reptilian immune systems have been rewired through alternative ligands, receptors, paralogous pathways, or regulatory architectures. Much like mammals that have lost key metabolic genes yet evolved novel strategies to fuel energetically demanding tissues, squamates may represent natural experiments in immune and developmental resilience. Studying how these animals maintain immune competence, development, and physiological stability across these conditions could reveal mechanisms of robustness and compensation that remain invisible in short-term laboratory knockouts. More broadly, the prevalence of gene loss in squamates contributes to an emerging inversion of a classic evo-devo theme: similar biological outcomes can be achieved even as the underlying genetic components change. Understanding how function persists despite “Key” gene loss may be as informative as discovering new genes themselves.
Critically, the confidence in such biological interpretations rests on rigorous methodology. Our framework provides essential quality control for genome assemblies and establishes a reproducible workflow for identifying lineage-wide gene losses with greater confidence.
Gene loss shapes evolution along two fundamental axes. First, it can be a direct agent of adaptive evolution [8], where the removal of a genetic constraint facilitates novel phenotypes—such as altered immune function or body plan reorganization. Second, it acts as a source of historical contingency, where the absence of a gene irrevocably alters a lineage’s genomic landscape, potentially creating evolutionary trade-offs, vulnerabilities, or unique dependencies that constrain future paths. Finally, these losses are not isolated events but clues to larger genomic dynamics. Just as chromosomal inversions can act as evolutionary "cassettes" for rapid adaptation [9], the segmental deletions and rearrangements that remove genes illustrate how genome architecture itself drives evolutionary change. The phylogenetic signal in these patterns of structural variation provides a unique window into genome evolution—a narrative that goes far beyond the story told by individual genes alone.
References
- Salve BG, Vijay N. Illuminating the mystery of thylacine extinction: a role for relaxed selection and gene loss. Proceedings of the Royal Society B Biological Sciences. 2025;292(2053):20251339. doi:10.1098/rspb.2025.1339
- Simões TR, Pyron RA. THE SQUAMATE TREE OF LIFE. Bulletin of the Museum of Comparative Zoology. 2021;163(2). doi:10.3099/0027-4100-163.2.47
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, et al. Towards complete and error-free genome assemblies of all vertebrate species. 2021;592:737–46.
- Kirilenko BM, Munegowda C, Osipova E, et al. Integrating gene annotation with orthology inference at scale. Science. 2023;380(6643):eabn3107. doi:10.1126/science.abn3107
- Sharma S, Shinde SS, Teekas L, Vijay N. Evidence for the loss of plasminogen receptor KT gene in chicken. Immunogenetics. 2020;72(9-10):507-515. doi:10.1007/s00251-020-01186-2
- Salve BG, Kurian AM, Vijay N. 2023 Concurrent loss of ciliary genes WDR93 and CFAP46 in phylogenetically distant birds. R. Soc. Open Sci. 10. (doi:10.1098/RSOS.230801)
- Salve BG, Sharma S, Vijay N. 2024 Evolutionary diversity of CXCL16-CXCR6: Convergent substitutions and recurrent gene loss in sauropsids. Immunogenet. 2024 , 1–19. (doi:10.1007/S00251-024-01357-5)
- Meier JI, McGee MD, Marques DA, et al. Cycles of fusion and fission enabled rapid parallel adaptive radiations in African cichlids. Science. 2023;381(6665):eade2833. doi:10.1126/science.ade2833
- Kirkpatrick M, Barrett B. Chromosome inversions, adaptive cassettes and the evolution of species’ ranges. Molecular Ecology. 2015;24(9):2046-2055. doi:10.1111/mec.13074
Follow the Topic
-
BMC Biology
This is an open access journal publishing outstanding research in all areas of biology, with a publication policy that combines selection for broad interest and importance with a commitment to serving authors well.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer metabolism
BMC Biology is calling for submissions to our Collection on Cancer metabolism. Cancer metabolism is a developing field that explores the biochemical and physiological changes that occur in cancer cells, which often exhibit distinct metabolic pathways compared to normal cells. Alterations such as the Warburg effect, where cancer cells preferentially utilize glycolysis for energy production even in the presence of oxygen, play a pivotal role in tumor progression and survival. Understanding the intricacies of cancer metabolism provides insights into how tumors adapt to their microenvironments and highlights potential therapeutic targets for intervention.
Future research in cancer metabolism promises to yield transformative insights that could reshape therapeutic approaches and improve precision medicine. The continued focus on metabolic pathways may lead to the identification of new biomarkers for cancer diagnosis and prognosis, as well as novel strategies to enhance the sensitivity of cancer cells to treatment. As we deepen our understanding of the metabolic landscape of tumors, we may uncover innovative strategies that exploit these vulnerabilities, ultimately leading to novel and more effective cancer treatments, as well as improved patient outcomes.
Recent advancements in the field, including the identification of metabolic reprogramming strategies and the influence of diet on tumor growth, have opened new avenues for research. Investigations into the roles of lipids, fatty acids, and dietary interventions, such as ketogenic diets, are revealing potential methods for manipulating tumor metabolism and enhancing the efficacy of existing treatments. We invite researchers to submit their work to this Collection, which aims to showcase groundbreaking research and technologies addressing cancer metabolism and support the advancement of this field, encompassing a wide array of topics related to metabolic pathways and their implications for cancer biology and therapy.
Potential topics include but are not limited to:
- Metabolic reprogramming in cancer cells
- The Warburg effect and its implications
- Role of mitochondria in cancer metabolism
- Impact of dietary interventions on tumor metabolism
- Glycolysis and lipid metabolism in cancer
- Animal models
- Imaging and method developments
- Metabolic engineering
This Collection supports and amplifies research related to SDG 3 (Good Health and Well-being).
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Oct 30, 2026
Biology of neurodegenerative diseases
BMC Biology is calling for submissions to our Collection on the biology of neurodegenerative diseases. This Collection aims to bring together multidisciplinary knowledge to better understand the mechanisms driving progressive neuronal dysfunction and loss.
We welcome studies investigating key pathological mechanisms, including protein misfolding and aggregation, neuroinflammation, oxidative stress, mitochondrial disorders, dysfunction of cellular protein sorting and degradation, altered RNA metabolism, blood-brain barrier impairment, brain vascular dysfunction, contribution of extracellular vesicles and synaptic dysfunction. Research exploring intracellular transport disruptions, neuronal network alterations, and genetic or epigenetic contributions to neurodegeneration is also encouraged.
We are particularly seeking submissions that employ state-of-the-art approaches, such as multi-omics, electrophysiology, high-resolution imaging, (induced) pluripotent stem cell-derived model systems, (e.g. microfluidics 2D co-culture, organoids and assembloids), and animal models to gain deeper mechanistic insights into neurodegenerative processes and identify potential therapeutic targets.
This Collection supports and amplifies research related to SDG 3: Good Health and Well-Being.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Sep 03, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in