Mechanical Activation of Noncoding RNA in Human Cardiomyocytes
Published in Bioengineering & Biotechnology
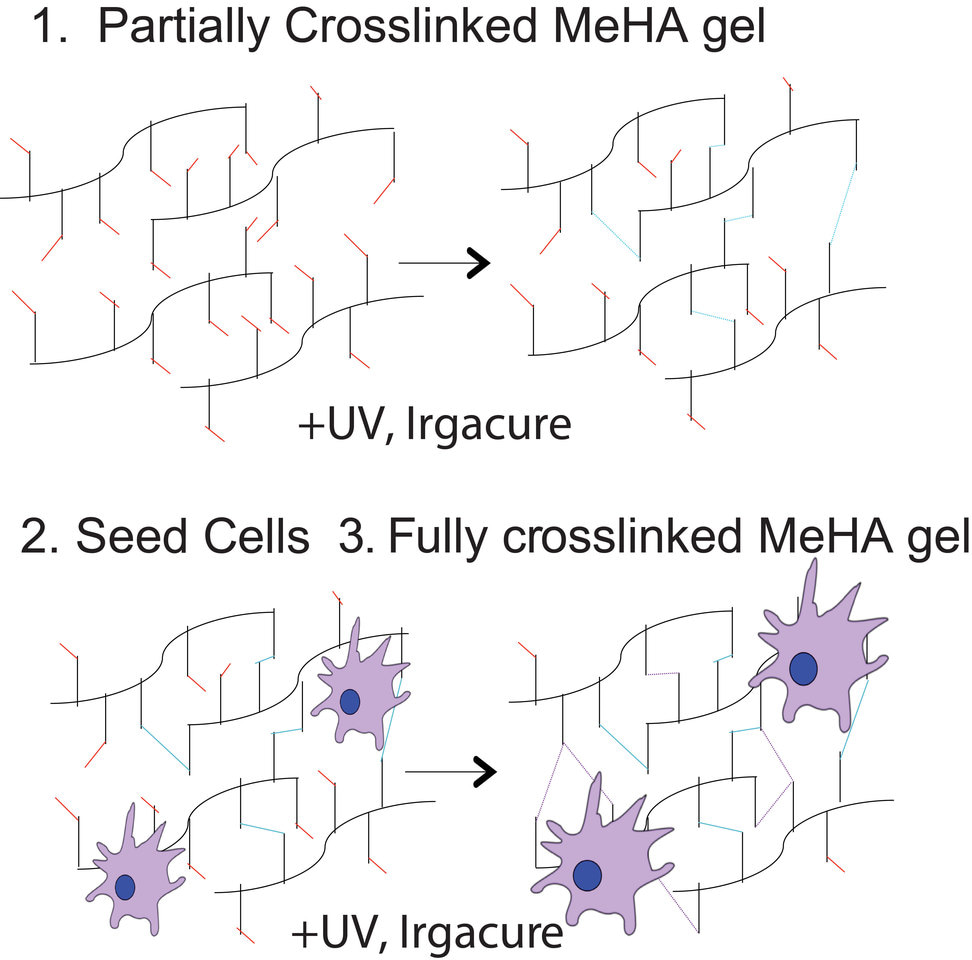

Despite the identification of millions of single nucleotide polymorphisms (SNPs) associated with disease, the molecular mechanisms of very few have been identified. This is because many SNPs are human-specific, non-coding, and context specific, e.g. needing specific stimuli to induce disease. During my Ph.D. work in the lab of Adam Engler, I focused on SNPs in the 9p21 gene locus, which represent the strongest genomic risk factor for heart disease, as a model locus in which to study the mechanisms of non-coding variants. In addition, to study these variants in the proper context, we utilized a dynamically stiffening methacrylated hyaluronic acid (MeHA) hydrogel that better mimic cardiac fibrosis.
We first began by developing the MeHA hydrogel such that it initially crosslinked into a soft matrix before in situ dynamic stiffening in the presence of cells, allowing cells to feel both “muscle-like” and “fibrotic-like” stiffness. Surprisingly, while cardiomyocytes with (R/R) or without (N/N) SNPs contracted synchronously in physiological niche, only R/R cardiomyocytes exhibited asynchronous contractions after stiffening. This result was confirmed in isogenic lines where the R/R locus was deleted (R/R KO). Given that gap junctions mainly regulate myocyte electrical conduction, we next investigated whether asynchronicity in R/R myocytes when cultures were dynamically stiffened was caused by alterations in expression of connexin 43, the most predominant gap junction protein found in ventricular cardiomyocytes. Indeed, we observed reduced connexin 43 mRNA and protein expression as well as reduced dye transfer in R/R cardiomyocytes after stiffening, indicating dysfunctional junctions.
To better understand the mechanisms involved, we first determined how the risk haplotype alters expression of the non-coding RNA ANRIL, which overlaps with the 9p21 gene locus and has been implicated in disease. We observed increased ANRIL expression in R/R cardiomyocytes compared to N/N and R/R KO cells. Next, given that ANRIL has been shown to silence the behavior of nearby cell cycle regulators that are associated with cellular stress responses, we measured p16 mRNA transcript and observed a marked up-regulation in response to stiffening in N/N and R/R KO cells but not R/R cells. We were next able to identify activation of the stress kinase c-Jun N-terminal kinase (JNK) in R/R cells after stiffening using a phospho-kinase screen, which was particularly intriguing as previous studies have suggested that p16 is capable of binding to JNK in response to stress and prevent its activation. We validated JNK activation in R/R cells by western blot and observed synchronous contractions, restored connexin 43 expression, and improved dye transfer when R/R cells were treated with a JNK inhibitor after stiffening. Thus, these data and literature together suggest a complex pathway whereby the risk haplotype increases expression of ANRIL to block p16 inhibition of JNK phosphorylation and transcriptional suppression of connexin 43.
Our data is supportive of a general approach where suitable biological and cell systems are developed to explore mechanism(s) in vitro and under pathologically appropriate mechanical stress when in vivo assays are impractical or impossible. This strategy can be broadly applied to study other SNPs while also highlighting the importance of studying these variants in the appropriate environmental context.
Follow the Topic
-
Nature Biomedical Engineering

This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.
Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Microphysiological systems for advanced modeling, high-throughput evaluation, and clinical translation
Publishing Model: Hybrid
Deadline: Dec 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in