

The concept of non-innocence has emerged as a promising platform for transition-metal catalysis.1-4 Originally introduced to describe the phenomenon of redox-active ligands in coordination compounds resulting in a formal oxidation state ambiguity of the central atom5, it equally captured the general notion that a chemical entity can have a secondary function apart from the canonically assigned one. This latter, broader conception build the foundation on which the concept of non-innocent electrophiles is based on. Looking at electrophiles from an user’s perspective, two basic primary functions can be attributed to them, i.e. providing an appropriate bond polarization and in most cases by that the localisation of the bond forming site (Figure 1a). However, working at the interface of organic chemistry and catalysis, I had come across literature reports in which electrophiles had an additional secondary function, and which was essentially a consequence of the corresponding leaving group (Figure 1b).6-9 These electrophiles can be considered non-innocent in comparison to conventional (innocent) electrophiles, e.g. organic halides or sulfonates, which only fulfil the above mentioned primary criteria and do not posses a secondary function.
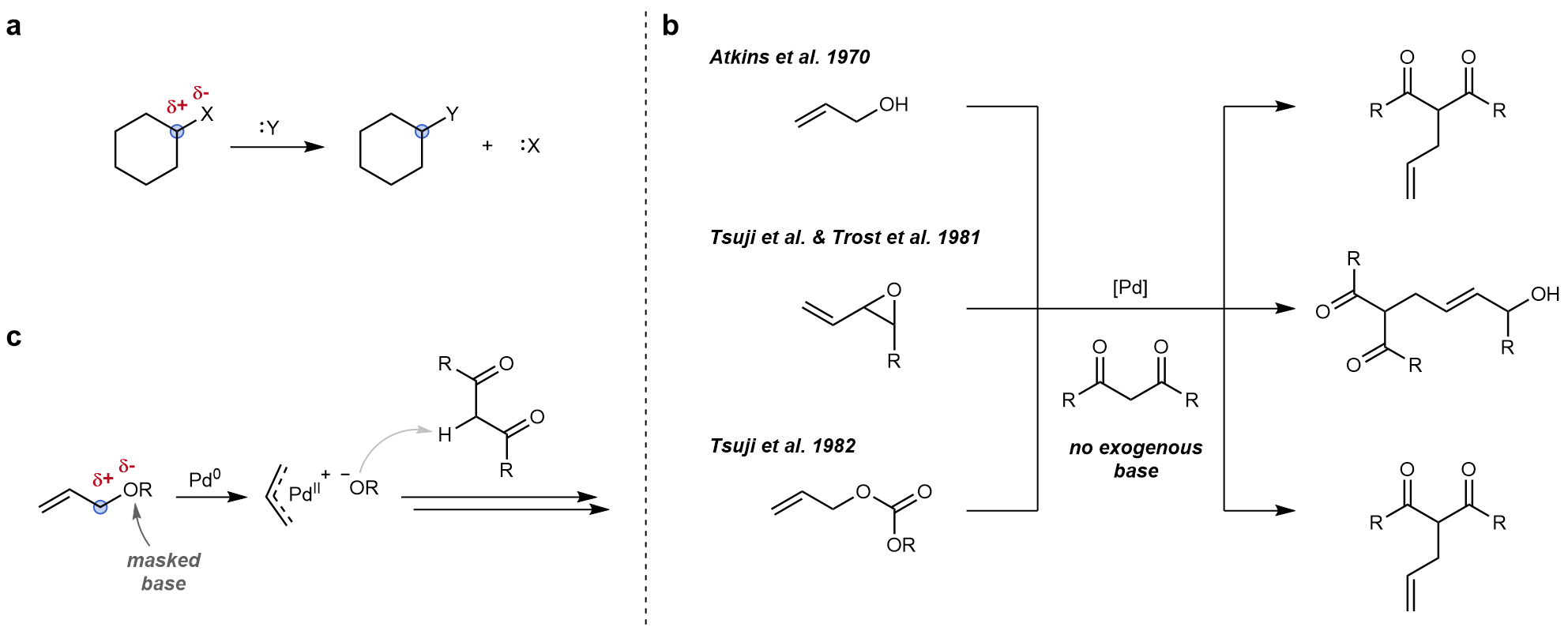
Figure 1 a: The two basic primary functions of electrophiles exemplified in a generic substitution reaction, i.e. an appropriate bond polarization (red) and the localisation of the bond forming site (blue). b: Seminal studies in the field of allylic substitution reactions that inspired the concept of non-innocent electrophiles. c: Allyl electrophiles carrying a masked base which is released upon oxidative addition.
In these seminal studies in the field of allylic substitution reactions, the strategic use of selected electrophiles enabled the authors to omit otherwise required stoichiometric amounts of exogenous base in such reactions. In all these cases the employed electrophile contains a masked base which is released upon oxidative addition (Figure 1c). Apart from not requiring the addition of any base, to me the more striking and ingenious feature was the linking of the base generation to an elementary step of the catalytic cycle leading to self-regulating catalytic systems that create the base only on demand. Having started my doctoral studies in 2018, I had witnessed several seminal reports dealing with the ‘base problem’ in cross-coupling chemistry that were published around that time.10-13 Inspired by the aforementioned allylic substitution reactions and equipped with a healthy dose of naivity as an early-stage researcher as well as the encouragement by my supervisor, I decided to try and extend this reactivity beyond the realm of allylic systems and apply it to tackle the ‘base problem’ in a general way. There had been some literature precedent suggesting that the envisioned reactivity could be viable and it quickly became apparent that the use of C–O electrophiles with strong C(sp2)–O bonds would most likely be key to achieving it.14,15 Luckily, I had been fortunate enough to conduct my master’s thesis in a leading group in the field of inert bond activation led by Prof. Dr. Ruben Martin at the ICIQ in Tarragona, Spain (although my actual thesis project dealt with carboxylation chemistry using CO2). Thanks to that I had already gained some experience in the field of Ni-catalysis and I directly knew that once more nickel would also be the metal of choice for this endeavor. As a consequence, a straightforward proof of principle study was compiled in which selected potentially non-innocent electrophiles, which might be able to release a base after oxidative addition, were reacted under prototypical C(sp2)–O bond activation conditions in three representative reactions in which at least one step requires the presence of a base, i.e. the Buchwald-Hartwig amination (BHA), the Mizoroki-Heck reaction (MHR) and the Suzuki-Miyaura coupling (SMC) (Figure 2).
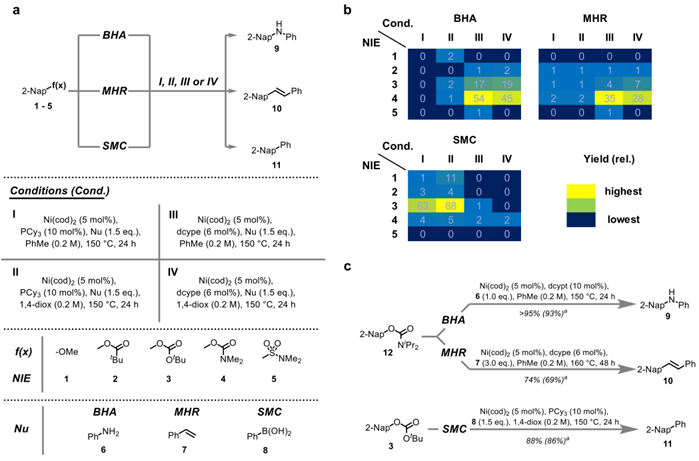 Figure 2 a: Proof of principle study with reaction conditions, selected potentially non-innocent electrophiles (NIE) and nucleophiles (Nu). b: Initial hit results with GC-yields. c: Optimized reaction conditions. aGC-yield (isolated yield in parentheses). cod, 1,5-cyclooctadiene; Cy, cyclohexyl; dcype, 1,2-bis(dicyclohexylphosphaneyl)ethane; dcypt, 3,4-bis(dicyclohexylphosphaneyl)thiophene; 1,4-diox, 1,4-dioxane; eq., equivalent(s); iPr, iso-propyl; Me, methyl; 2-Nap, 2-naphthalenyl; Ph, phenyl; PhMe, toluene; rel., relative; tBu, tert-butyl.
Figure 2 a: Proof of principle study with reaction conditions, selected potentially non-innocent electrophiles (NIE) and nucleophiles (Nu). b: Initial hit results with GC-yields. c: Optimized reaction conditions. aGC-yield (isolated yield in parentheses). cod, 1,5-cyclooctadiene; Cy, cyclohexyl; dcype, 1,2-bis(dicyclohexylphosphaneyl)ethane; dcypt, 3,4-bis(dicyclohexylphosphaneyl)thiophene; 1,4-diox, 1,4-dioxane; eq., equivalent(s); iPr, iso-propyl; Me, methyl; 2-Nap, 2-naphthalenyl; Ph, phenyl; PhMe, toluene; rel., relative; tBu, tert-butyl.
This study afforded lead results and demonstrated that the envisioned concept was indeed able to promote the studied couplings in the absence of added base. After optimization diisopropylcarbamates as well as tert-butyl carbonates were found to release a competent base after oxidative addition – a strategy which proved to be generally applicable. As a result multiple coupling reactions (8 distinct classes of coupling reactions) which traditionally rely on the addition of (super)stoichiometric base could be turned into exogenous base-free, homogeneous processes, that were compatible with base-sensitive functional groups.
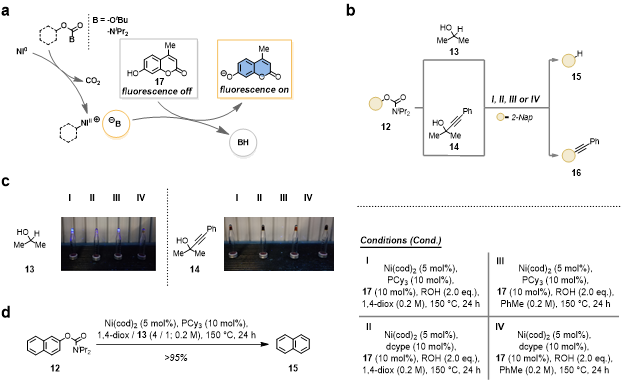
Figure 3 a: Design principle for the fluorescence-based assay. b: Outline of the micromole-scale assay and the investigated reactions and conditions. c: Results of the assay based on the visual inspection of the samples after irradiation at 366 nm. d: Discovered deoxygenation reaction with optimized conditions.
In addition, the advantageous features of non-innocent electrophiles over conventional electrophiles were demonstrated in multiple applications, such as reaction miniaturization, reactivity sensing and reaction discovery. This led to the development of a micromole-scale fluorescence-based assay for reaction discovery that reliably and rapidly detects reactivity using minimal amounts of materials (Figure 3). Importantly, the assay does not require customized substrates or specialized equipment as it makes use of an inexpensive, exogenous fluorescent sensor, i.e. 4-methylumbelliferone 17, which is employed in substoichiometric amounts (10 mol%) and only requires a common benchtop UV-lamp for visualization of the reaction outcome. Using the developed assay, a Ni-catalyzed deoxygenation reaction of aryl carbamates using isopropanol as a benign reductant was discovered.
For more details especially on important prior studies in the field of C–O bond activation and Ni-catalysis, without which this work would not have been possible please have a look at our article:
https://www.nature.com/articles/s41929-022-00770-x
References
- Chirik, P. J., Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 327, 794–795 (2010).
- Praneeth, V. K. K., Ringenberg, M. R., Ward, T. R. Redox-Active Ligands in Catalysis. Angew. Chem. Int. Ed. 51, 10228–10234 (2012).
- Lyaskovskyy, V., de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2, 270–279 (2012).
- Luca, O. R., Crabtree, R. H. Redox-active ligands in catalysis. Chem. Soc. Rev. 42, 1440–1459 (2013).
- Jørgensen, C. K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy. Coord. Chem. Rev. 1, 164‒178 (1966).
- Atkins, K. E., Walker, W. E. & Manyik, R. M. Palladium catalyzed transfer of allylic groups. Tetrahedron Lett. 11, 3821–3824 (1970).
- Tsuji, J., Kataoka, H., Kobayashi, Y. Regioselective 1,4-addition of nucleophiles to 1,3-diene monoepoxides catalyzed by palladium complex. Tetrahedron Lett. 22, 2575–2578 (1981).
- Trost, B. M. & Molander, G. A. Neutral alkylations via palladium(0) catalysis. J. Am. Chem. Soc. 103, 5969–5972 (1981).
- Tsuji, J., Shimizu, I., Minami, I. & Ohashi, Y. Facile palladium catalyzed decarboxylative allylation of active methylene compounds under neutral conditions using allylic carbonates. Tetrahedron Lett. 23, 4809–4812 (1982).
- Cox, P. A. et al. Base-catalyzed aryl-B(OH)2 protodeboronation revisited: from concerted proton transfer to liberation of a transient aryl anion. J. Am. Chem. Soc. 139, 13156–13165 (2017).
- Chen, L., Sanchez, D. R., Zhang, B. & Carrow, B. P. ‘Cationic’ Suzuki–Miyaura coupling with acutely base-sensitive boronic acids. J. Am. Chem. Soc. 139, 12418–12421 (2017).
- Malapit, C. A., Bour, J. R., Brigham, C. E. & Sanford, M. S. Base-free nickel-catalysed decarbonylative Suzuki–Miyaura coupling of acid fluorides. Nature 563, 100–104 (2018).
- Dennis, J. M., White, N. A., Liu, R. Y. & Buchwald, S. L. Breaking the base barrier: an electron-deficient palladium catalyst enables the use of a common soluble base in C–N coupling. J. Am. Chem. Soc. 140, 4721–4725 (2018).
- Wang, Y., Wu, S.-B., Shi, W.-J. & Shi, Z.-J. C‒O/C‒H coupling of polyfluoroarenes with aryl carbamates by cooperative Ni/Cu catalysis. Org. Lett. 18, 2548–2551 (2016).
- Nishizawa, A. et al. Nickel-catalyzed decarboxylation of aryl carbamates for converting phenols into aromatic amines. J. Am. Chem. Soc. 141, 7261–7265 (2019).
Follow the Topic
-
Nature Catalysis

This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in