'Peroxisomal β-oxidation acts as a sensor for intracellular fatty acids and regulates lipolysis
Published in Healthcare & Nursing
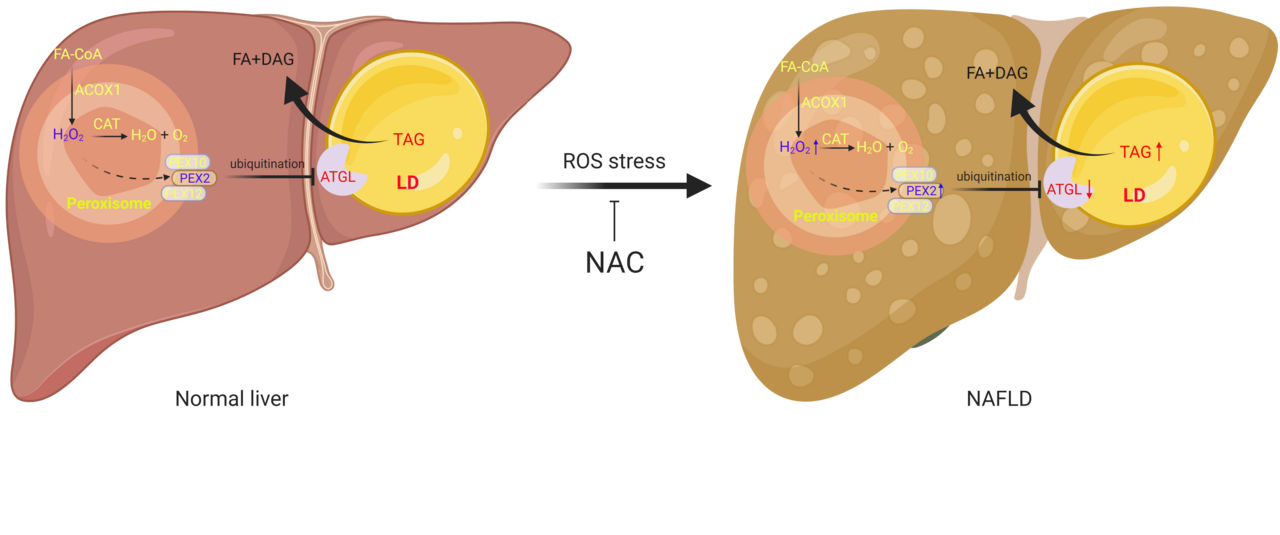
Peroxisomes are widely abundant organelles with diverse functions, such as β oxidation of very long chain fatty acid (VLCFA), branched chain fatty acid (BCFA), ROS clearance and synthesis of bile acids and ether lipids, to name but a few. For the past decades, the intimate physical interaction between lipid droplets (LDs) and peroxisomes was observed in various cell types, however, the physiological relevance of these contacts has remained elusive. Recent publications could show that fatty acid, liberated by lipolysis can be directly taken up by peroxisomes1. Besides, it was shown that PEX5, the key factor mediating the import of peroxisomal matrix proteins, can interact with the lipase ATGL to promote the recruitment of the latter to lipid droplet surface in adipocytes upon hormone stimulation2. Nevertheless, it is intriguing to understand the relationship, between peroxisomal activity and LD lipolysis. Therefore, we wanted to investigate whether different peroxisomal metabolic processes could impact on fatty acid release by regulating LD lipolysis (https://www.nature.com/articles/s42255-021-00489-2).
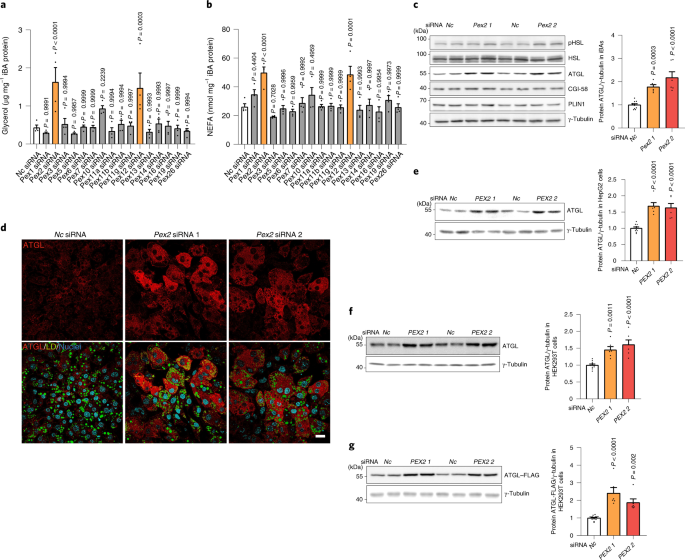
To this end, we utilized initially brown adipocytes, which exhibit abundant peroxisomes and LDs in close contact. Quite unexpectedly, we observed a significant upregulation of lipolysis and ATGL levels upon depletion of PEX2/10/12 in a small-scale screen targeting all peroxins. Moreover, we could show that this regulation was not limited to adipocytes, but consistent in other cell types. PEX2 functions as a RING-containing E3 ligase and we could demonstrate that peroxisomes in contact with LDs function as platform for the regulation of LD surface distributed lipase ATGL through PEX2.
Interestingly, the protein levels of PEX2 are kept at very low levels in cells3. Hence, we postulated there might be a regulatory mechanism which links PEX2 levels to peroxisomal activity. Given that peroxisome dependent oxidation of VLCFA, generates H2O2, we were first curious whether PEX2 could be stabilized by peroxisomal ROS. Should this be the case, the whole process would constitute a feedback mechanism to modulate intracellular fatty acid levels based on the activity of peroxisomal β oxidation. In line with our hypothesis, we could demonstrate that PEX2 was indeed stabilized by peroxisomal ROS or exogenous H2O2 due to the intramolecular and intermolecular disulfide bridges within PEX2. Moreover, we found that alterations of peroxisomal ROS regulates ATGL levels and lipolysis through this PEX2 dependent mechanism. These findings suggest a broader substrate specificity in peroxisomal β oxidation as VLCFAs are not the only fatty acid species released by lipolysis and indeed we could show that different fatty acids with different carbon lengths could regulate lipolysis via the aforementioned pathway.

Lipotoxicity 0is believed to promote disease progression in the context of the metabolic syndrome. Hence, we argued that an existence of a cell autonomous mechanism regulating fatty acid overload could impact on cell lipid homeostasis. Indeed, we could show that downregulation of peroxisomal ROS via genetic ablation of hepatic ACOX1 or through systemic administration of antioxidants reduced triglyceride levels in liver, while CAT depletion increased peroxisomal ROS, which in turn led to triglyceride accumulation. These processes were dependent on the presence of the components of the newly identified pathway demonstrating that hepatic peroxisomal ROS levels can indeed regulate ATGL levels and TG deposition, and thus might serve as a novel target to prevent liver steatosis. Our further work based on human liver samples suggests that steatosis levels are positively correlated with ROS levels but negatively correlated with ATGL levels. Recent work showed that both peroxisomal β oxidation and ATGL overexpression can induce lipophagy in liver, which would pose an alternate way to mobilize triglyceride4,5. How peroxisomal β oxidation, ATGL mediated lipolysis and lipophagy cooperate precisely to control intracellular fatty acid homeostasis is an interesting question to be explored.
- Chang, C.-L. et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J Cell Biol 218, 2583–2599 (2019).
- Kong, J. et al. Spatiotemporal contact between peroxisomes and lipid droplets regulates fasting-induced lipolysis via PEX5. Nature Communications 11, 1–16 (2020).
- Sargent, G. et al. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J Cell Biol 214, 677–690 (2016).
- He, A. et al. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Molecular Cell (2020) doi:10.1016/j.molcel.2020.05.007.
- Sathyanarayan, A., Mashek, M. T. & Mashek, D. G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Reports 19, 1–9 (2017).
Follow the Topic
-
Nature Metabolism

This journal publishes work from across all fields of metabolism research that significantly advances our understanding of metabolic and homeostatic processes in a cellular or broader physiological context, from fundamental cell biology to basic biomedical and translational research.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in