PhyloVelo enhances transcriptomic velocity field mapping using monotonically expressed genes
Published in Genetics & Genomics
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular diversity and dynamics in development and disease. However, scRNA-seq data only capture a snapshot of gene expression at a given time point, which limits our ability to understand the past and future cell states and trajectories. RNA velocity, a computational method that leverages the kinetics of spliced and unspliced RNAs, has been a powerful tool to overcome this limitation and predict the direction and speed of cell-fate transitions. However, RNA velocity relies on several assumptions that may not hold in complex biological systems. Moreover, RNA velocity may be affected by technical limitations such as the sparsity and bias of scRNA-seq data.
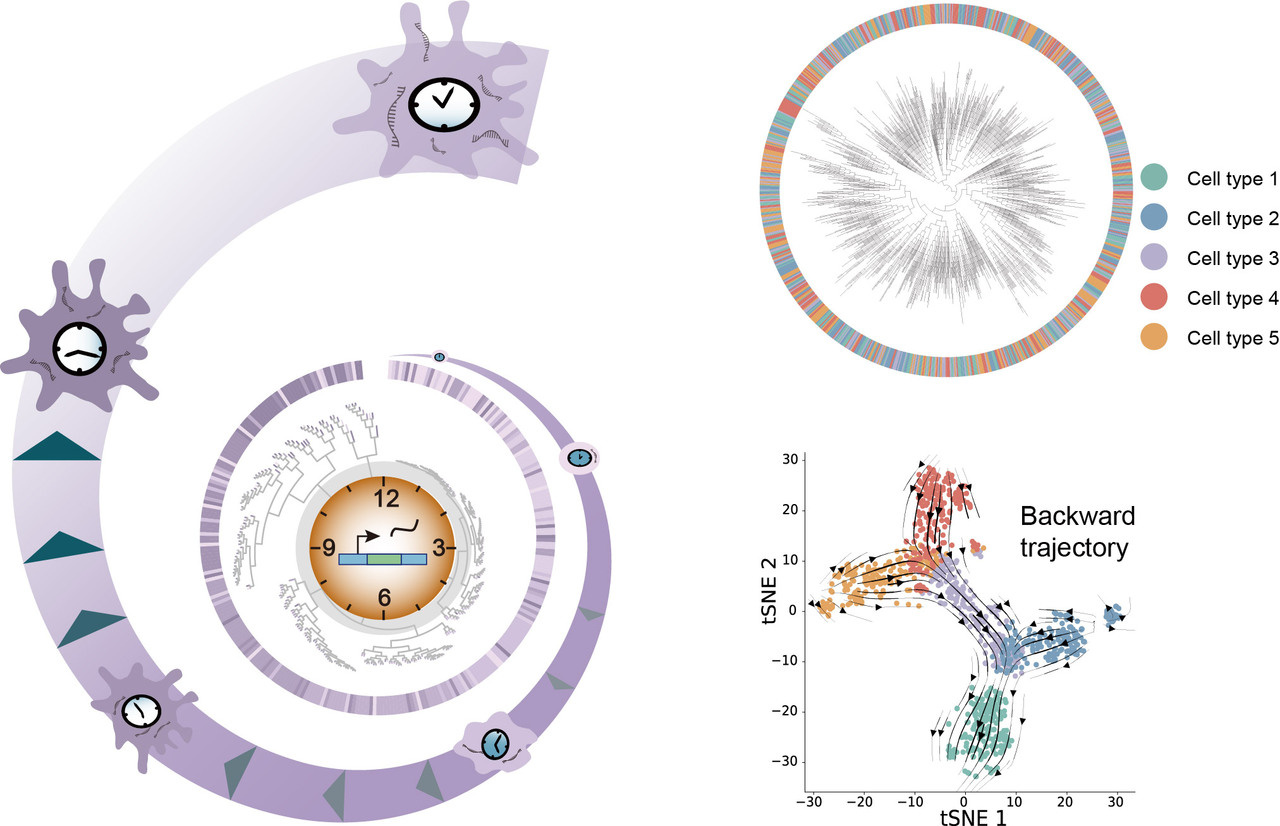
In our study, we introduce PhyloVelo, a novel computational framework that estimates the velocity of transcriptomic dynamics by using monotonically expressed genes (MEGs): genes that have expression patterns that either increase or decrease, along the phylogenetic time of single cells. Phylogenetic time is defined as the number of cell divisions or mutations that separate a cell from a common progenitor. By integrating scRNA-seq data with lineage information, such as CRISPR/Cas9-based lineage tracing or static barcoding, PhyloVelo identifies MEGs and reconstructs a transcriptomic velocity field that points to the past states of cells on low-dimensional embedding. PhyloVelo can be applied to diverse biological contexts such as normal development, tumor evolution and immune response.
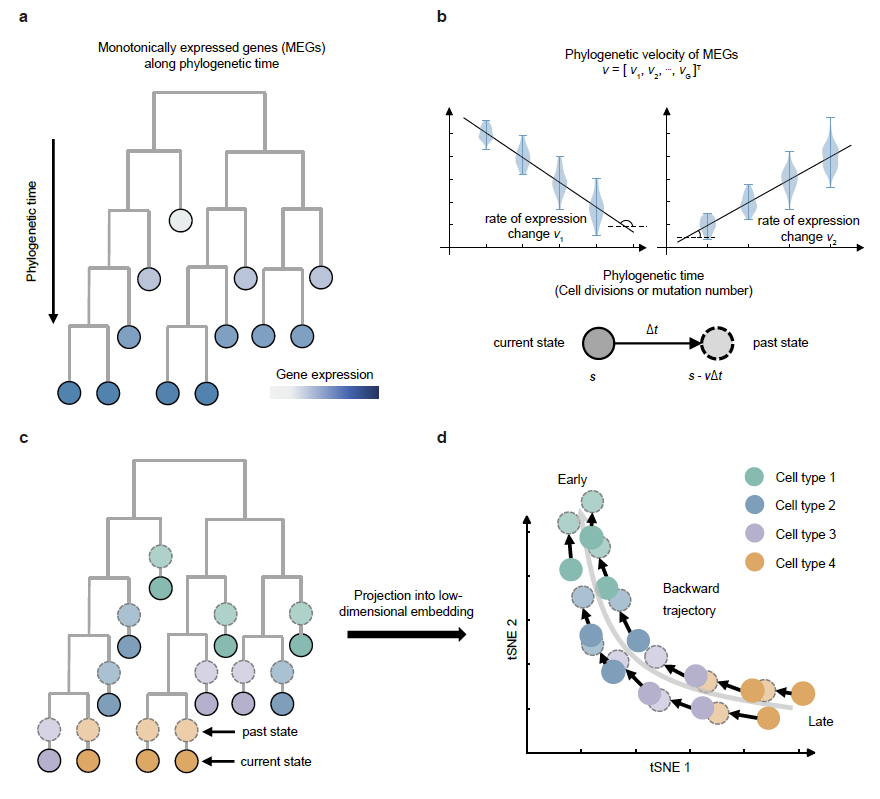
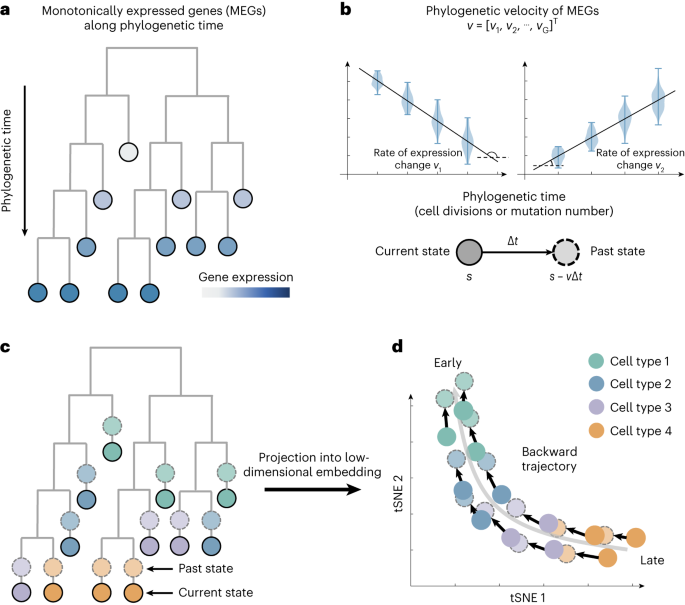
Fig. 1. Schematic of the PhyloVelo framework. (a) Schematic of monotonically expressed genes (MEGs) over phylogenetic time on a cell phylogenetic tree. (b) Two examples of MEGs whose latent expressions are associated with the phylogenetic time. (c) Phylogenetic velocity predicts the past transcriptional state of a cell before a unit of phylogenetic time. (d) Projection of the phylogenetic velocity into low dimensional embedding enables the mapping of cell-state trajectory in backward directions.
The idea of PhyloVelo originated from an observation that cells with closer lineage relationships usually have similar expressions for some genes, even if their cell types may differ. Therefore, we made a reasonable assumption that gene expression patterns can be divided into two types: cell type-related genes and cell type-unrelated genes, and the cell type-unrelated genes are highly correlated with their lineage relationships, among which genes with specific expression patterns (such as MEGs in this article) can serve as “clocks” to indicate the developmental state of cells. However, gene expression data usually have high noise, which makes it difficult for these “clock” genes to be identified. To this end, we use zero-inflated negative binomial distribution, negative binomial distribution, and Gaussian distribution to denoise gene expression data, and the gene expression after denoising is called potential expression.
To test our hypothesis, we used a diffusion process with drift to model the variation of latent expression of each gene with phylogenetic time, and estimated its drift coefficient as the velocity of the gene. The velocity field of cell differentiation can be constructed by accurately identifying enough MEGs. To test the model, we first applied PhyloVelo to simulation data where various lineage structures were considered, including linear, bifurcated and convergent differentiations. We found that PhyloVelo could robustly identify MEGs and accurately recover the expected trajectories in all scenarios. We also benchmarked PhyloVelo with real lineage tracing data from C. elegans embryos, where the invariant lineage tree is entirely known. We found that PhyloVelo outperformed RNA velocity in recapitulating the actual developmental orders.
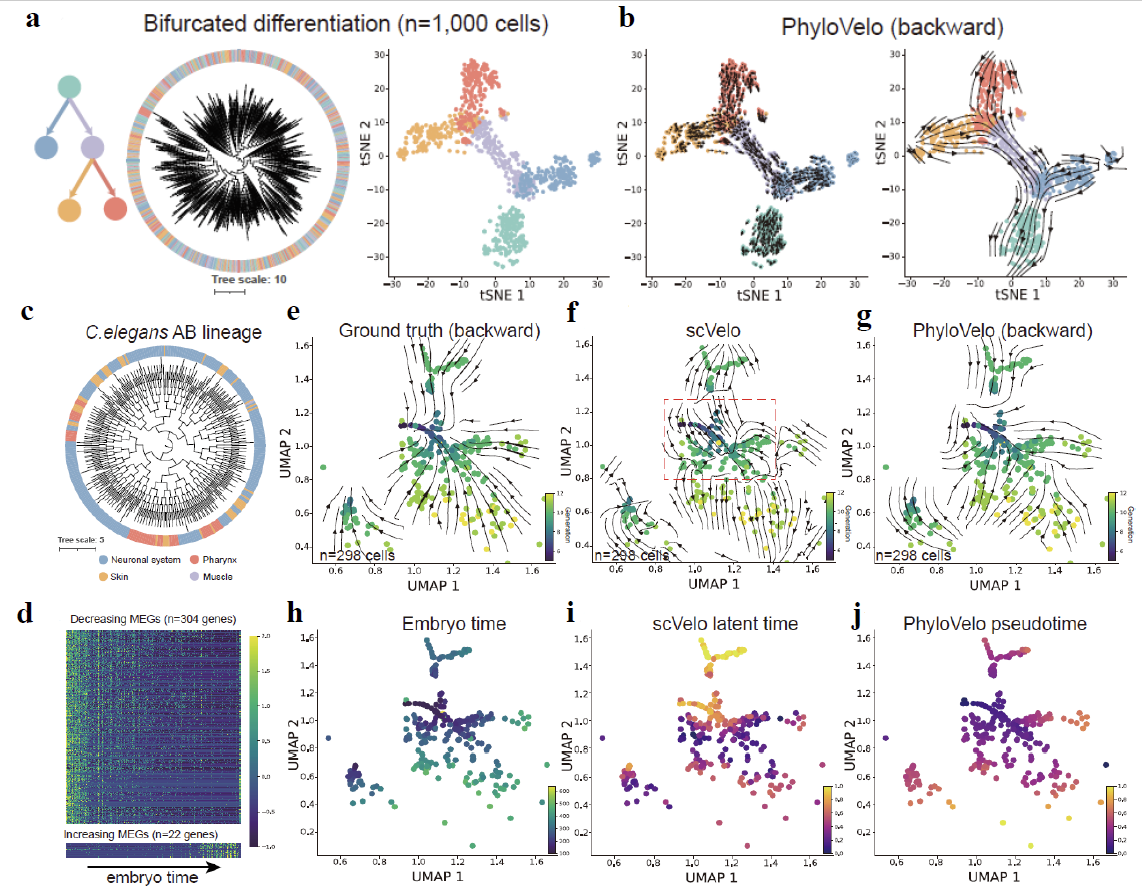
Fig. 2. PhyloVelo recovers complex cell lineages in simulations and C. elegans. (a) Simulation of single-cell RNA-seq data and coupled cell-division history under bifurcated differentiation model. (b) Phylogenetic velocity fields reconstructed by PhyloVelo for the corresponding differentiation scenarios. (c) Phylogenetic tree of the C. elegans AB lineage. (d) Heatmap showing the expressions (z-score normalized) of MEGs along C. elegans embryo time. (e) The ground-truth velocity fields where vectors are superimposed on the cells that point to each of their immediate parental cells on the UMAP plot. (f-g) The velocity fields estimated by scVelo (f) or PhyloVelo (g). Dash square indicates the early embryonic lineages where RNA velocity gave erroneous estimations on the fate directions. (h) C. elegans embryo time as Packer et al., 2019. (i) scVelo latent time. (j) PhyloVelo pseudotime.
We then applied PhyloVelo to several CRISPR/Cas9-based lineage tracing datasets from mouse or human, including mouse embryo development, in vitro hematopoiesis, lung tumor evolution and intratumoral T cell dynamics. We demonstrated that PhyloVelo could resolve complex lineage structures and quantify state-transition probabilities between cell types. We also showed that PhyloVelo could circumvent the multiple-rate kinetics issue of RNA velocity and transfer MEGs across independent datasets in similar biological conditions. Interestingly, we found that MEGs across tissues and organisms were strongly enriched in ribosome-mediated processes, suggesting an internal clock-like gene expression program during cell proliferation and differentiation.
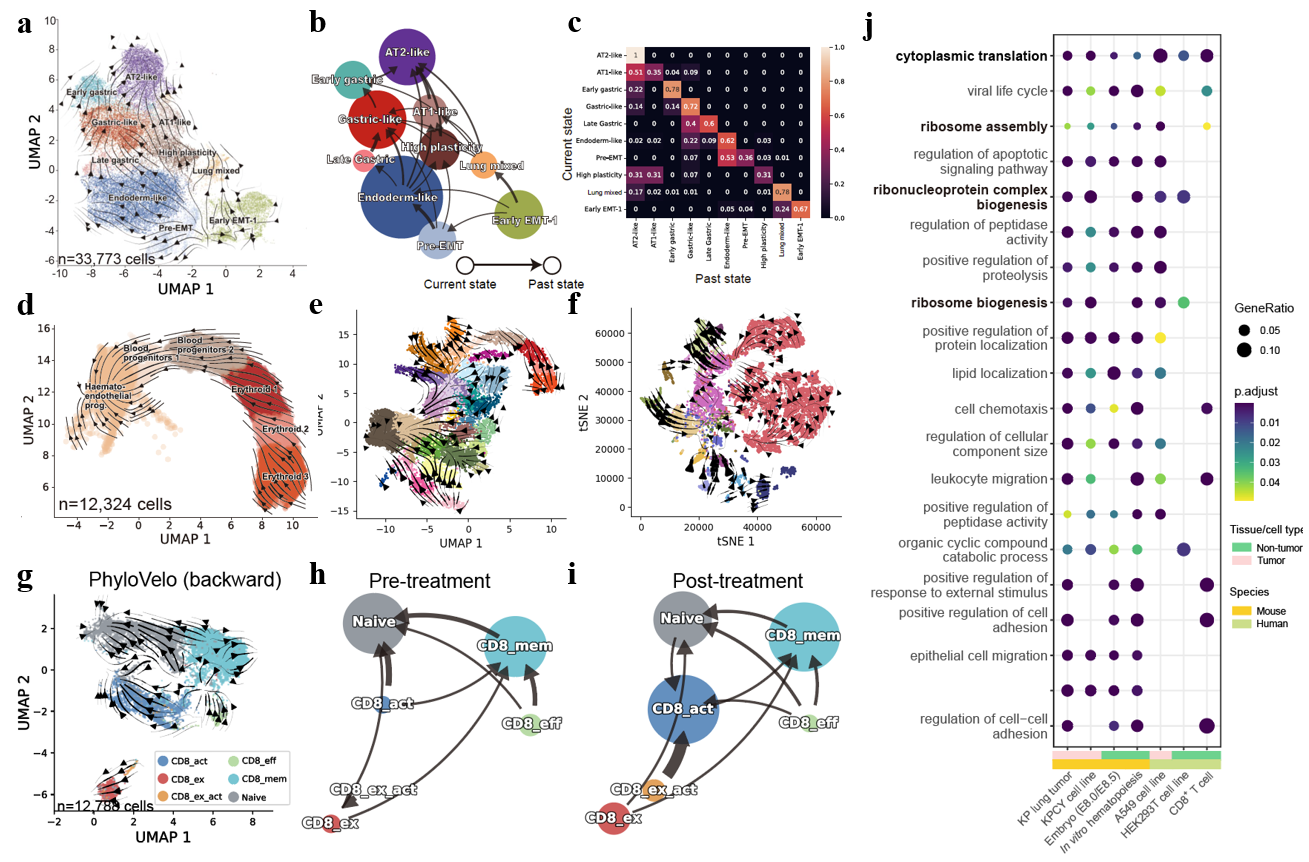
Fig. 3. PhyloVelo enables cell fate analysis for complex biological processes. (a-c) PhyloVelo velocity fields (a), cell-type transition graph (b) and cell-type transition matrix (c) for 33,773 single cells from mouse lung adenocarcinomas (Yang et al., 2022). (d, e) PhyloVelo velocity fields of erythroid maturation (d) and whole mouse embryos (e) datasets from Pijuan-Sala et al., 2019. (f) PhyloVelo velocity fields of mouse brain development from La Manno et al., 2021 (g) The velocity fields of tumor-infiltrating CD8+ T cells in BCC samples estimated by PhyloVelo. Data were from Yost et al., 2019. (h, i) Cell-type transition graph (backward) at pre-treatment (h) and post-treatment (i). (j) Gene ontology (GO) enrichment of MEGs identified across tissues and organisms.
Some of the challenges we faced during this project were related to the quality and availability of single-cell lineage tracing data. For example, we had to deal with low coverage and high dropout rates of scRNA-seq data, which could affect the identification of MEGs and the estimation of phylogenetic time. We also had to account for different sources of noise and variation in lineage tracing methods, such as off-target effects of CRISPR/Cas9 editing or stochasticity of barcode evolution. To address these issues, we used very stringent conditions to screen MEGs. We also used simulated data to show that PhyloVelo is still very robust under lower quality datasets.
We believe that PhyloVelo provides a novel perspective and a complementary approach to RNA velocity for studying cellular dynamics from lineage-resolved scRNA-seq data. By exploiting both transcriptomic and lineage information, PhyloVelo can reveal hidden features of cell fate transitions that are otherwise difficult to capture by gene expression alone. We hope that PhyloVelo will facilitate new discoveries in developmental biology and disease progression.
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in