Real-time Evolution: Monitoring SARS-CoV-2 Mutations via the PED Algorithm
Published in Microbiology, Genetics & Genomics, and Biomedical Research

The novel coronavirus (Severe Acute Respiratory Syndrome Coronavirus 2, SARS‑CoV‑2), first identified in Wuhan at the end of 2019, spread rapidly worldwide by January 2020. In Japan, infections were initially reported among people who had dined together on traditional houseboats and among cruise ship passengers, eventually developing into a full-scale pandemic.
SARS‑CoV‑2 shares similarities with the earlier SARS‑CoV outbreak; however, the amino acid sequence of the spike (S) protein responsible for receptor binding differs substantially. Although both viruses use ACE2 as the cellular entry receptor, the mode of interaction with ACE2 is different in SARS-CoV-2. This resulted in markedly increased binding affinity and infection efficiency for SARS‑CoV‑2.
The more stable binding to the ACE2 protein enabled efficient early replication in airway epithelial cells, leading to extremely high transmissibility. In addition, the high frequency of asymptomatic infections meant that infected individuals often continued normal activities, facilitating explosive global spread.
Although the case fatality rate of SARS‑CoV‑2 is considered lower than that of SARS‑CoV, its high transmissibility led to a massive number of infections. As a result, many people—particularly the elderly and those with compromised immune systems—lost their lives.
The nucleotide sequence of SARS‑CoV‑2 was released at a very early stage by Chinese researchers. A distinctive feature for research on this pandemic was the rapid and wide public availability of next‑generation sequencing (NGS) data derived directly from patient samples. In particular, the United Kingdom analysed sequencing data from a very large number of patients, which became available for download from the NCBI Sequence Read Archive (SRA).
We analysed these downloaded sequences using our virus-tailored modification of PED program to detect genetic variants. By applying PED’s bidirectional alignment method, we were able to efficiently detect both single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) in various sequenced virus genome samples.
PED includes a function that determines homozygous and heterozygous states based on the frequency of detected variant candidates. However, this framework is not appropriate for SARS‑CoV‑2, which is not a diploid organism. In practice, SARS‑CoV‑2 samples often contained mixed infections of multiple variants, and the observed variant frequencies varied widely among mutations.
To address this, we extended PED by adding a function that outputs read counts for each detected variant, specifically targeting organisms like SARS‑CoV‑2 that do not exhibit homozygous or heterozygous genotypes. This enhancement enabled more appropriate confirmation of viral mutations.
In our study, we downloaded virus genome sequences form ca. 50 individuals per sampling date and detected polymorphisms by our modified PED. During the early stages of the pandemic. As there were many days with fewer than 50 reported, we analysed all available data for those days.

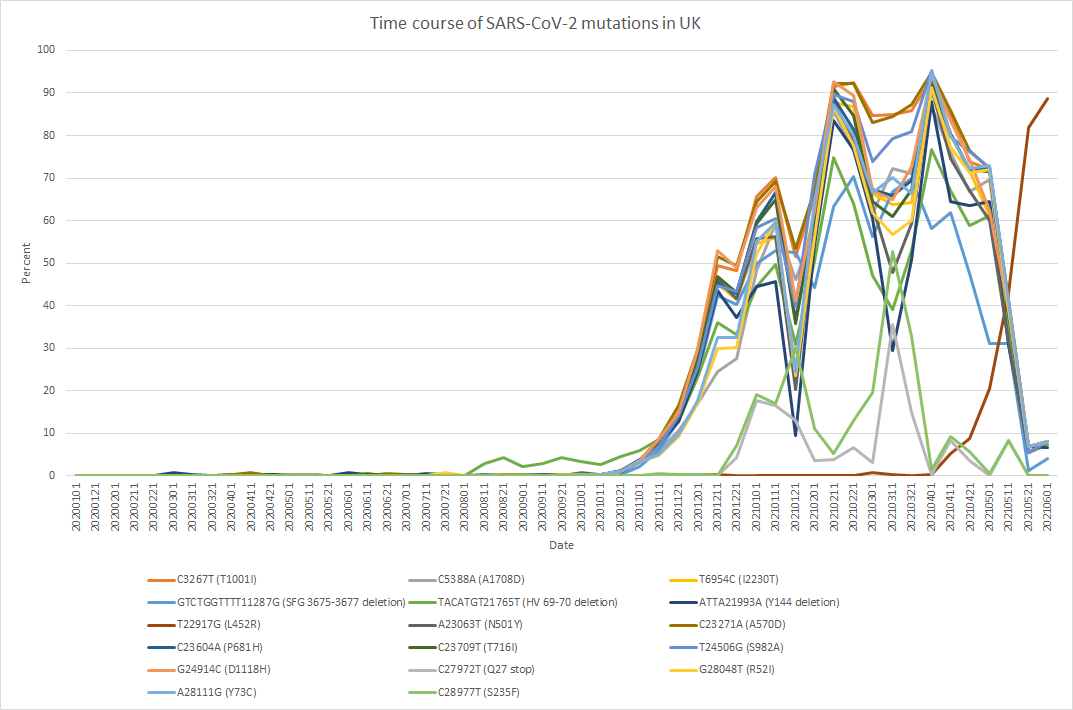
The figure shows an example of the detection frequency of mutations plotted at monthly intervals from the start of the pandemic. From 2020 until around May 2021, the Alpha variant carrying the N501Y (A23063T) mutation was dominant, after which it was replaced by the Delta variant carrying the L452R (T22916G) mutation. We also obtained extensive information on insertion and deletion mutations, which are a particular strength of PED. Although PED was originally developed primarily for detecting polymorphisms in eukaryotic organisms, it produced results for viral variants that were largely consistent with previously reported findings. When NGS data were available, results could be obtained within just a few minutes.
In this way, we were able to observe the evolution of SARS‑CoV‑2 during the pandemic almost in real time. However, eventually we suspended this monitoring analysis because SRA later limited downloads of the metadata for sample collection dates to approximately 100,000 records, preventing access to more recent data.
The ability to analyse mutations directly from publicly available raw sequencing data—without waiting for expert consensus analyses—was a highly significant development made evident during this pandemic.
Follow the Topic
-
BMC Bioinformatics

This is an open access, peer-reviewed journal that considers articles describing novel computational algorithms and software, models and tools, including statistical methods, machine learning and artificial intelligence, as well as systems biology.
Related Collections
With Collections, you can get published faster and increase your visibility.
Predictive toxicology
BMC Bioinformatics is welcoming submissions to our Collection on Predictive toxicology.
Predictive toxicology investigates the harmful effects of chemical substances using models and data-driven methods, often aiming to decrease dependence on traditional animal testing, such as mammals, for assessing health risks. Developments in this field support New Approach Methodologies (NAMs) for evaluating chemical safety and regulation. NAMs refer to any methods that enhance safety assessments while avoiding animal testing. Specifically, predictive toxicology employs computational techniques with a mechanistic understanding of toxicity to estimate risks to human health and the environment.
Recent advances have highlighted the use of various technologies that generate data valuable for in silico toxicity prediction, including omics, in vitro screening, high-throughput phenotyping, organoids, and alternative in vivo models. These innovations, combined with comparative biology and insights from other disciplines (e.g., genetics, evolution), refine hazard and risk assessment methods, facilitating a more precise evaluation of chemical safety and ultimately improving health outcomes.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for predictive toxicology.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Aug 14, 2026
Cell tracking
BMC Bioinformatics is welcoming submissions to our Collection on Cell Tracking.
Cell tracking is a technique used to monitor and analyze the movement and behavior of cells over time, allowing the study of cellular behaviors, dynamics, and interactions within various biological contexts. Advanced bioinformatics tools play a vital role in analyzing cell tracking data. They help identify cell movement patterns and understand their biological implications. These tools are particularly relevant when processing large datasets and when investigating cell cycles.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for cell tracking. We encourage contributions detailing methods for detecting and characterizing cell movements to better understand cell migration and behavior.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Jun 23, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in