Recovering counts from proportions
Published in Protocols & Methods and Statistics

To understand biological systems, we are able to make measurements of different classes of molecules: proteins, transcripts, metabolites, etc. However, these measurements are often missing an important piece of information.
While biomolecules are always present as counts (e.g. 1,137 EGF protein molecules in a cell), many measurement modalities are limited to discerning proportions (e.g. 1 out of 1,000,000 protein molecules are EGF). In these cases, there is a single scalar missing from the measurement: the total number of proteins.
This gap between our measurement and the biological state can happen for multiple reasons. The most common is 'non-exhaustive sampling'. For instance, a transcriptomic protocol samples reads from a pool of RNAs, and the sequence of the read is used to discern which transcript it corresponds to. Since not every transcript in the original pool was drawn, the output does not contain explicit information about total number of transcripts, and thus the results (after various processing steps) are compositional.
However, there may be a way to get count information out of the proportional result, if used in conjunction with the right prior.
Using the information from proportions
What information do proportions contain? While they don't directly relate to the total count, they do restrict what combinations of component counts are possible. For instance, if we observe that 1/100 reads map to transcript A, and 2/100 map to transcript B, we know in terms of possible counts, 50 of A and 100 of B is feasible, while the reverse is not.
The question then becomes: Over what space are proportions an informative constraint? The answer is not the space of possible total counts, which is just one dimension, but the multi-dimensional space of combinations of component counts (i.e. counts of protein A, protein B, protein C). The proportion can be thought of as a vector that cuts through this space, restricting possible count values to a line.
So the prior should be constructed over the component count space, essentially encoding how likely combinations of component counts (e.g. 50 of protein A and 40 of protein B) are. While many choices of probability distribution for the prior are possible and reasonable, we used the multivariate normal distribution, as it can account for covariance while remaining simple and easy to work with.
Once these two pieces (the proportion vector and the prior) are in place, we are then able to calculate the posterior distribution of counts. We utilize the simple relation that a proportion times the total count is equal to the component count. This is the reverse of very common practice of dividing component counts by the total to get proportions or percentages.
Using this relation, we can establish a posterior probability distribution solely as a function of the total count (given the proportion vector and the prior). From the posterior we can then get our estimate of the total count present, and confidence intervals. We named this process Mahalanobis Count Inference (MCI), after the mathematician Prasanta Chandra Mahalanobis, who did pioneering work on multivariate normal distributions that was key to MCI's derivation.

Moving into log-space
While we can conduct this procedure on counts in linear space, something interesting happens when we do the same in log-space (i.e. log-transforming all counts and the proportion vector).
Moving to log-space is motivated by two observations: 1) while counts cannot be negative, the linear model assigns a non-zero probability to negative counts; and, 2) many biological counts tend to vary multiplicatively rather than additively, and are thus better fit by log-normal distributions. Taking the log of the counts and proportion vector resolves both of the above issues. But something else emerges as a mathematical consequence of the model.
In linear space, all possible proportion vectors intersect at zero, and then go off in their specified direction. When you transform proportion vectors to log-space their point of intersection ceases to exist (as the log of 0 is undefined). After transformation, all vectors point in the same direction, and are therefore parallel.
Interestingly, all parallel one-dimensional cross sections of a multivariate normal distribution have the same variance. For us, this means that all possible proportion vectors will have the same-sized confidence intervals in log-space, for a given prior. We can calculate these confidence intervals with just the covariance matrix of the prior.
Why does this matter? Well, since we want the count inference to be accurate, we want the confidence intervals to be as small as possible. We can use this to evaluate how well a prior will work without needing to test it empirically. This allows us to easily distinguish and choose between possible priors without testing them on proportions.
Constructing references
Importantly, not all features that are present in the measurement and composition (e.g. Protein A-Z) need to be represented in the prior. Any subset will do, and the best priors retain the features with the most stable counts. The above method in log-space allows this to be done quantitatively, directly, and without needing to test predictions empirically.
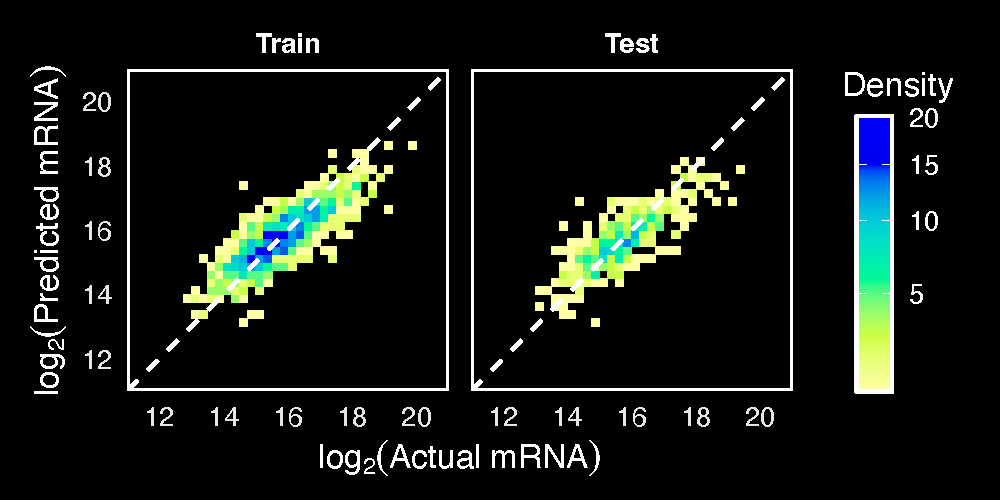
The MCI procedure is agnostic to the measurement type (e.g. proteomics, transcriptomics, etc.), and could in principle be applied in any proportional situation if its assumptions are met. Doing so would require the construction of a reference which is applicable to the biological situation. In our manuscript, we constructed references to predict white blood cell counts from cell proportions and total mRNA within single cells.
The method requires known counts of features in the training data to construct the prior, so some method for measuring counts initially is necessary. After training, only proportions are necessary. In single-cell transcriptomics, known quantities of spike-in RNAs (ERCCs) can be used. It is also key that the molecules of interest have a sensible and consistent container. For the case of bulk RNA, the total transcript count will be strongly confounded by the amount of tissue added, and thus training a reference on these bulk settings is not advisable. Conversely, applications to single-cell analyses are particularly attractive.
The future
As we were able to infer total mRNA counts quite well (r=0.81), it raises the possibility that MCI references could be applied to various single-cell settings. Though importantly, the cells used for training and testing in the manuscript were all a similar cell type. It remains to be shown if this approach could work for many diverse cell types.
We also found that existing single-cell transcriptomic analysis methods (scran, TMM) do not appear to capture count variation between cells (see Supplementary Figure 3 in manuscript). These methods seem to have the opposite implicit assumptions as MCI. Where MCI recapitulates biological variation in total counts, most existing workflows (outside of spike-factor normalization) treat this variation as technical. This further points to potential value in developing MCI references in this single-cell transcriptomics.
Outside of building references, more intricate architectures utilizing MCI may be possible. Perhaps references trained on single-cell data may be applicable to bulk data, with predictions interpreted as the counts of an 'average cell'. Single-cell references may also be applied to spatial applications for count inference, and could conceivably be used for partitioning spatial data into cells. For more diverse biological settings, perhaps classification schemes into multiple references could be used.
Alongside these more speculative applications, the most concrete major step is building references for applications of interest, such as single-cell proteomics and transcriptomics. We hope that researchers in the community will use their expertise to build references for various questions of interest to them. Please reach out if you are interested in building a reference, as we would be happy to provide advice, collaborate, or answer any questions.
Follow the Topic
-
npj Systems Biology and Applications
An online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach and encourages studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems.

Related Collections
With Collections, you can get published faster and increase your visibility.
Systems mechanobiology
Publishing Model: Open Access
Deadline: Sep 28, 2026
Systems immunology: multi-omics approaches, dynamical modeling and novel agentic AI approaches
Publishing Model: Open Access
Deadline: Sep 12, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in