Reprogramming Mitochondrial Metabolism to Overcome AML Drug Resistance: TMQ0153 as a Promising Therapeutic Candidate
Published in Cancer, Cell & Molecular Biology, and Pharmacy & Pharmacology
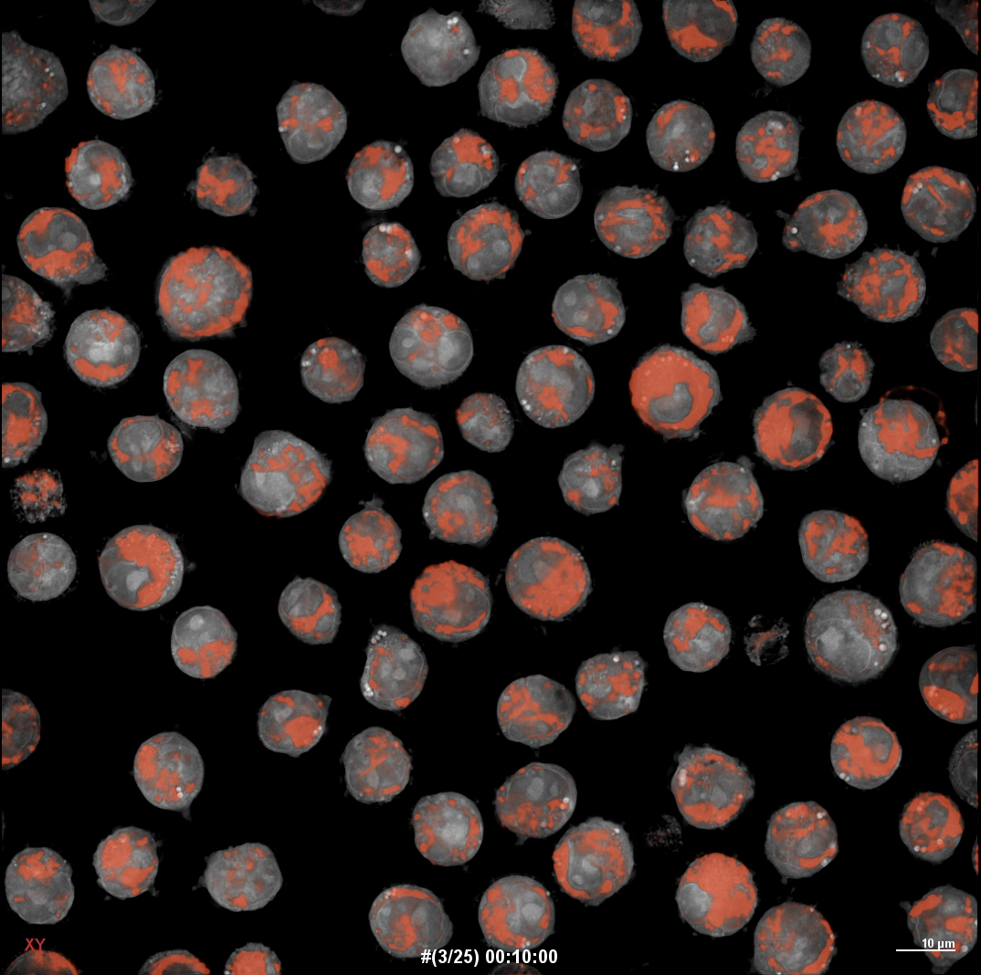
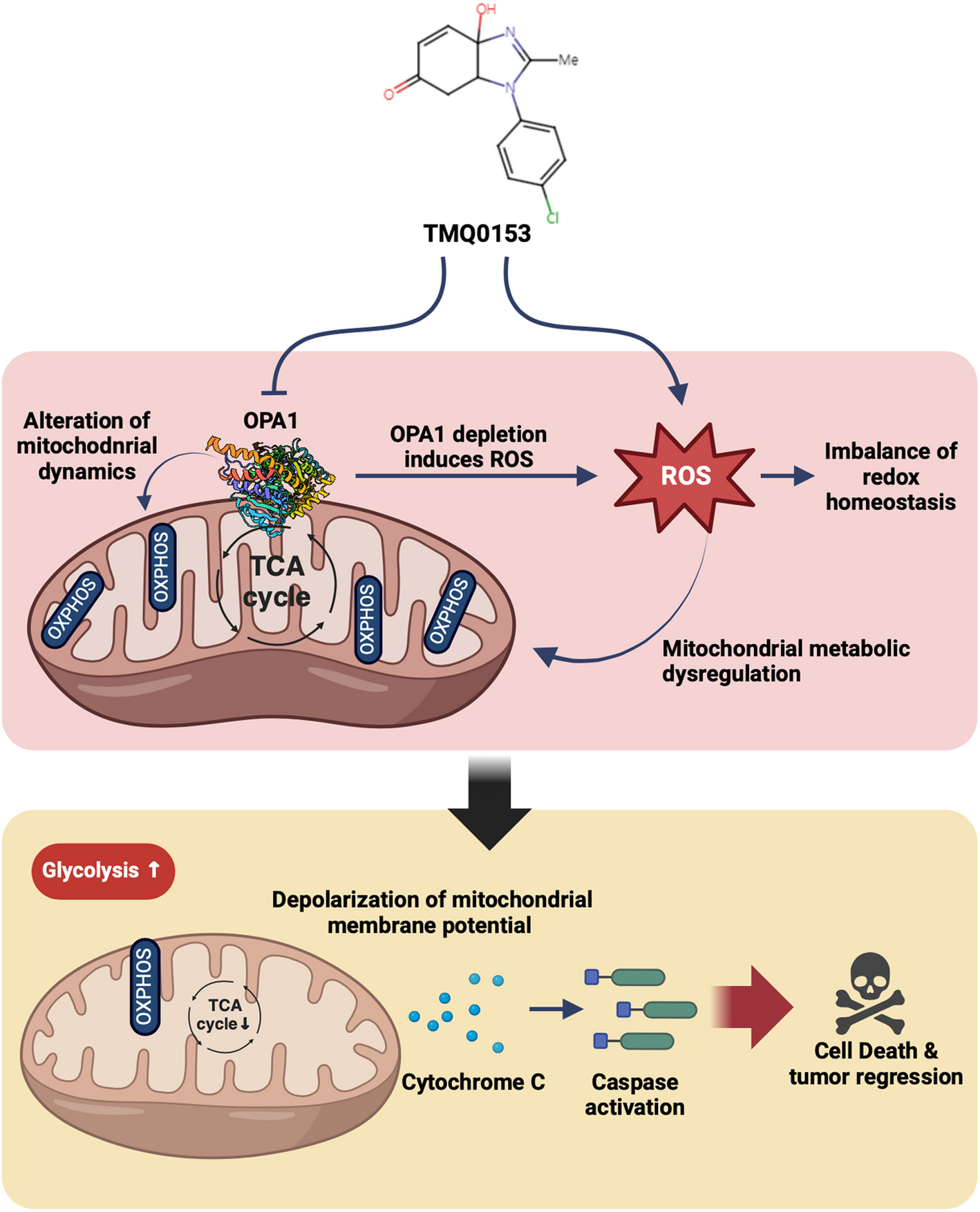

Acute myeloid leukemia (AML) remains a challenging malignancy to treat, particularly due to its aggressive nature, high relapse rates, and persistent drug resistance. One hallmark of resistant AML is its reliance on mitochondrial fitness and altered energy metabolism to support survival and evade therapy 1. In this context, our recent preclinical work introduces TMQ0153 2, a tetrahydrobenzimidazole derivative, as a potent disruptor of mitochondrial homeostasis and a promising adjuvant to existing treatment modalities 3.
We previously examined TMQ0153 for its potential anti-cancer effects on chronic myeloid leukemia (CML) cells 4. Prior findings indicated that TMQ0153 disrupts mitochondrial function and increases oxidative stress by elevating ROS levels and promoting the accumulation of cytoplasmic Ca²⁺. Additionally, we observed decreased intracellular glutathione (GSH) levels during the early stages of TMQ0153-induced autophagy and necroptosis in CML cells. The change in oxidative stress levels caused by TMQ0153 affects mitochondrial homeostasis, leading to various forms of cell death, including immunogenic cell death (ICD), a chemotherapy-induced tumor cell death. These findings suggest that TMQ0153 is a promising agent for targeting mitochondria. Next, we investigated TMQ0153, focusing on mitochondrial metabolism and dynamics in AML.
TMQ0153 exerts its anti-leukemic activity by selectively targeting the mitochondrial fusion protein OPA1, which is often upregulated in AML cells with adverse risk profiles. Inhibiting OPA1 leads to disrupted mitochondrial dynamics, loss of cristae integrity, and mitochondrial membrane depolarization. These structural disruptions are accompanied by a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis, elevated ROS levels, and marked apoptotic cell death.
Metabolically, TMQ0153 significantly alters intracellular redox states, as reflected by changes in the NAD⁺/NADH and GSH/GSSG ratios, underscoring its role in inducing oxidative stress-mediated cytotoxicity. In vivo, TMQ0153 reduces tumor burden in xenograft models and demonstrates synergistic potential when combined with clinically relevant agents such as gilteritinib and venetoclax/azacitidine. These combination regimens prolong survival and reduce leukemic load more effectively than monotherapies alone.
TMQ0153 offers a novel mechanism to resensitize AML cells to treatment by targeting mitochondrial vulnerability and reprogramming leukemia cell metabolism. These findings open new avenues for developing metabolism-targeted combination therapies in AML, particularly in resistant or high-risk patient populations. Ongoing studies will further explore its clinical translation and potential in personalized AML therapy.

A mechanistic overview describing the effect of TMQ0153.
Created in BioRender. Park, S and Diederich, M. (2025)
References:
- Cerella C, Dicato M, Diederich M. BH3 Mimetics in AML Therapy: Death and Beyond? Trends Pharmacol Sci 2020;41(11):793-814. doi: 10.1016/j.tips.2020.09.004 [published Online First: 20201005]
- Tran MQ, Nguyen TB, Sawadogo WR, et al. Unaromatized Tetrahydrobenzimidazole Synthesis from p‐Benzoquinone and N‐Arylamidines and their Cytotoxic Potential. European Journal of Organic Chemistry 2018;2018(42):5878-84. doi: 10.1002/ejoc.201801077
- Park SJ, Cerella C, Kang JM, et al. Tetrahydrobenzimidazole TMQ0153 targets OPA1 and restores drug sensitivity in AML via ROS-induced mitochondrial metabolic reprogramming. J Exp Clin Cancer Res 2025;44(1):114. doi: 10.1186/s13046-025-03372-0 [published Online First: 20250407]
- Song S, Lee JY, Ermolenko L, et al. Tetrahydrobenzimidazole TMQ0153 triggers apoptosis, autophagy and necroptosis crosstalk in chronic myeloid leukemia. Cell Death Dis 2020;11(2):109. doi: 10.1038/s41419-020-2304-8 [published Online First: 20200207]
Follow the Topic
-
Journal of Experimental & Clinical Cancer Research
Journal of Experimental & Clinical Cancer Research is an online peer-reviewed journal that publishes original research papers, reviews and commentaries in cancer research, from bench to bedside.

Related Collections
With Collections, you can get published faster and increase your visibility.
Insights into the relationship between metabolism and cancer
Cancer metabolism has become one of the most studied aspects of the "hallmarks of cancer." It refers to alterations in how tumor cells process nutrients to fuel their rapid proliferation and spread. These dramatic dysregulations include elevated glucose uptake even in the presence of oxygen (the Warburg effect), dependence on glutamine, and the production of waste products such as lactate, which further support tumor growth and metastasis. Genetic mutations accumulating during cancer progression in metabolic enzymes and cancer-promoting signaling pathways directly affect metabolic changes in cancer cells as well as in the tumor microenvironment [1,2].
A growing number of scientists are developing targeted therapies to exploit these metabolic differences to slow or halt cancer progression. Some therapies aim to block or alter the production of specific metabolites, while several drugs for other pathologies, such as diabetes, are of great interest due to their potential to slow tumor growth by altering systemic metabolic factors such as insulin and glucose levels in a "drug repositioning" strategy [3-7].
Cancer metabolism, involving altered lipid, iron, and amino acid pathways, regulates ferroptosis, an iron-dependent, lipid peroxidation-driven form of cell death. Cancer cells often resist this process, but targeting their specific metabolic vulnerabilities, such as glutathione depletion or iron overload, can trigger ferroptosis to kill tumor cells and overcome drug resistance [8].
Cancer cells can rewire their metabolism when one pathway is blocked, so it is becoming clear that combining metabolism inhibitors with traditional chemotherapy, radiotherapy, or immunotherapy may be an effective therapeutic strategy [8]. While some metabolism-targeting drugs have been successful (e.g., in Acute Lymphocytic Leukemia), targeting cancer metabolism is complex due to its similarity to normal cell processes [9]. Metabolic profiling of patients is helping to identify specific vulnerabilities, leading to more personalized metabolic therapies [10].
Understanding cancer metabolism at a deeper mechanistic level is also essential to improving immunotherapy, as immune cell metabolism changes during therapy administration, contributing to acquired resistance.
KEYWORDS: metabolomics; proteomics, genomics, mitochondrial metabolism, ferroptosis, oxidative stress, inflammation, innovative study models, new translational therapeutic approaches.
In this Collection of the Journal of Experimental & Clinical Cancer Research, we would like to promote studies that further dissect the molecular mechanisms through which cancer cells reprogram their own metabolism and that of cells in the tumor microenvironment, thus driving disease progression. Furthermore, studies that highlight potential metabolic vulnerabilities that could be targeted therapeutically will be of interest.
References
- Finley LWS. What is cancer metabolism? Cell. 2023;186(8):1670-88. PubMed PMID: 36858045. PMCID: PMC10106389.eng
- Guertin DA, Wellen KE. Acetyl-CoA metabolism in cancer. Nat Rev Cancer. 2023;23(3):156. PubMed PMID: 36658431. PMCID: PMC11137663.eng
- Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671-84. PubMed PMID: 21878982.
- Zou W, Han Z, Wang Z, Liu Q. Targeting glutamine metabolism as a potential target for cancer treatment. J Exp Clin Cancer Res. 2025;44(1):180. PubMed PMID: 40598593. PMCID: PMC12210561.
- Formenti L, Abramo F, Dellavedova G, Dematteis V, Decio A, Grasselli C, et al. Impairment of oxidative metabolism compromises Rad51 recruitment and potentiates PARP inhibitor effectiveness in ovarian cancer. J Exp Clin Cancer Res. 2026;45(1):45. PubMed PMID: 41530766. PMCID: PMC12888472.
- Chen T, Xu ZG, Luo J, Manne RK, Wang Z, Hsu CC, et al. NSUN2 is a glucose sensor suppressing cGAS/STING to maintain tumorigenesis and immunotherapy resistance. Cell Metab. 2023;35(10):1782-98.e8. PubMed PMID: 37586363. PMCID: PMC10726430.
- Pillai U J, Ray A, Maan M, Dutta M. Repurposing drugs targeting metabolic diseases for cancer therapeutics. Drug Discov Today. 2023;28(9):103684. PubMed PMID: 37379903.
- Wu Y, Li H, Yue K, Jing C, Duan Y. Ferroptosis in cancer: metabolism, mechanisms and therapeutic prospects. Mol Cancer. 2025;24(1):303.
- Punnasseril JMJ, Auwal A, Gopalan V, Lam AK, Islam F. Metabolic Reprogramming of Cancer Cells and Therapeutics Targeting Cancer Metabolism. Cancer Med. 2025;14(18):e71244. PubMed PMID: 40956032. PMCID: PMC12439291.
- da Silva-Diz V, Herranz D. Unleashing the Full Potential of Metabolic Interventions in T-ALL. Blood Cancer Discov. 2025;6(3):163-7. PubMed PMID: 40111138. PMCID: PMC12050939.
- Benjamin DI, Cravatt BF, Nomura DK. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab. 2012;16(5):565-77. PubMed PMID: 23063552. PMCID: PMC3539740.
Publishing Model: Open Access
Deadline: Feb 28, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in