Seeking antibiotic resistance dynamics among patient MICs in ATLAS
Published in Microbiology

(Here is our paper. Apologies about the acronyms!)
It's hard to read a discussion, a blog, a textbook or review about the increasing availability of data in biology and medicine without encountering some reference to Artificial Intelligence, Machine Learning, their manifold clustering, classification and decision algorithms.
And one area where doctors could do with some help is the problem of antibiotic resistance.
So is it too forlorn, or even foolish, a hope that AI, in all its guises, could help clinicians make better antibiotic treatment decisions, or design more highly optimised antibiotic stewardship practices? It seems a reasonable enough question, but Big Data Analytics, or AI, or whatever it's called nowadays, needs data, and lots of it. Usually the more the better.
Clinical microbiology labs generate lots of data - antibiograms, antibiotic susceptibility tests (ASTs), disk diffusion methods, the Biomerieux Vitek II machines and broth dilution - whatever the terminology and methodology you prefer - on a global scale, these labs generate thousands and thousands of MIC values on a daily basis.
MIC - the minimal inhibitory concentration gives clinicians some idea of how a microbe responds to an antibiotic in the lab and different agencies do curate large databases of MICs, but some are more useful than others for the purposes I have in mind.
The database belonging to the organisation EUCAST has many thousands of MIC datapoints, although it has no metadata of the type that an AI methodology could get its teeth stuck into. Similarly, the ESPAUR report made available in the UK only reports on frequencies of resistance - these are helpful for tracking changes in resistance but are of fairly limited value to AI algorithms that we might like to use to seek better ways of working with antibiotics.
However, as a group of quantitative and clinical microbiology researchers, our interest was piqued when Pfizer, Micron, Wellcome and the Open Data Institute published ATLAS. This database, curated by Micron, holds raw MIC data assembled across many years for thousands of patients and hundreds of pathogen-antibiotic combinations. So we downloaded the spreadsheet, with some glee, and set about coding in C++, Python and Matlab.
At this point, I would like to say that our first coding escapade was a rip-roaring success. But it wasn't. Sure enough, machine learning-style methods could find patterns, linkages and correlations between resistance in one place and another and between one bug and another, but so what? That's what our reviewers said: we don't like these data, they really should not be out there in this unfettered form and you haven't even checked for bias. Reject.
Bias. *&$!. Yes, we hadn't. And that's despite co-author, Jess, telling us to.
It was a concern those reviewers said MIC databases shouldn't even be in the public domain - which has also been said by colleagues - but it's hard to see how having less data can lead to greater understanding.
So, ploughing on, we looked for bias. And, as with many studies in the field, the pitfalls are obvious: the data derive from disk diffusion, broth dilution, Vitek machines, and on it goes ... so many methodologies all in one dataset. Then, of course, data are not replicated because these are clinical antibiotic susceptibility tests. And the US supplies much more data to ATLAS than does Africa, who supply hardly any. And some pathogens are far more represented than others, and data quantity shifts markedly in some specific years. And so on, such is the scope for bias. Pfizer were also much more interested in resistance to one or two particular antibiotics, so patient sampling bias is more likely than not.
At this point we need to explain something about MICs. We don't analyse completely raw MIC data but, rather, we transform them by first dividing MIC values by a published CLSI breakpoint and thereafter we take logs (i.e. log base 2). This means that 0 in all our figures below marks the dividing line between what is deemed resistant by CLSI, and what is not. Accordingly, negative data correspond to MICs that are below the CLSI breakpoints, whereas positive ones are above CLSI breakpoints.
Returning to the matter at hand, we came up with Python code to detect the potential for bias in ATLAS and the following figure shows just what can go wrong. We found this by writing code that looked for year-year correlation matrices in MIC distributions that have a "block" structure, just as you can see in 2 of the greeny-yellowy patchwork matrices on the left side in the figure below:

The key point here is sub-figure B: this shows a histogram of all the log2 MIC values in ATLAS for the antibiotic clindamycin when used against Streptococcus pneumoniae. The interesting thing about that sub-figure is the apparent reduction of a highly resistant cluster of Strep within that histogram around 2010, when its log2 MIC dropped at around 10 units and a new cluster appeared around 5 units, as if out of nowhere.
We first thought this was evidence of some kind of rapid evolutionary dynamic, but it isn't. It turns out that ATLAS is formed of 2, or so, different sub-databases so we separated them out into distinct datasets and analysed each one in turn. This lead to a very different picture.
The rightmost plots in sub-figures C and D now show what the 2 databases say: this pathogen-antibiotic combination has 2 MIC distributions that look very stable to the eye, where both are formed from clusters with both negative and position log2 MIC values on the x-axis. All this means that the merging of 2 sub-databases created what looks to the untrained eye like an evolutionary dynamic but which is, in fact, an artifactual dataset, as shown in sub-figure B.
Despite problems like this, ATLAS is an amazing resource, almost one of a kind, and we are grateful to all concerned for having the courage to put it out there, warts and all. It would be churlish to not dig deeper into what ATLAS has to say. So, we sought more anomalies like the above, uncovering over 100 possible cases, and we used some machine learning ideas (really little more than the statistical modelling of shifts of Gaussian clusters) to track how MIC distributions are changing through time.
These ideas lead us to something we call the R-MIC in our paper. This is the mean MIC of the cluster identified in the MIC histogram using Gaussian mixture modelling that, at a given time, has the largest MIC. The following figure illustrates this idea when we're looking 'down' onto an MIC distribution from above. In this particular case there are two clusters in the MIC distribution at all times, one with drug-resistant MIC values and one with drug-susceptible values.
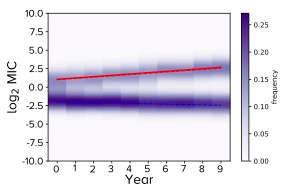 The R-MIC for a given year is the y-axis value of the red line and its slope is the yearly change in R-MIC. Note how the two clusters are moving apart whereby the R-MIC is increasing and the MIC of the rest of the distribution is slightly decreasing. Notice also how this is reminiscent of 'disruptive selection' from evolutionary genetics theory.
The R-MIC for a given year is the y-axis value of the red line and its slope is the yearly change in R-MIC. Note how the two clusters are moving apart whereby the R-MIC is increasing and the MIC of the rest of the distribution is slightly decreasing. Notice also how this is reminiscent of 'disruptive selection' from evolutionary genetics theory.
Given features like this, we felt that the mean MIC is not the best statistic to track resistance and, instead, we determined the R-MIC and predicted its changes over the coming years for several important pathogens for all the antibiotics in ATLAS. This is what we found, where 'MIC' now means R-MIC:
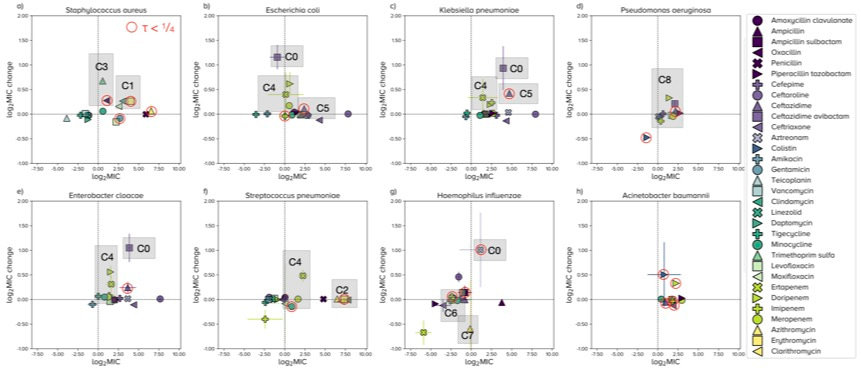
In one sense, the most problematic cases here are those dots where log2 MIC is close to the y-axis and yet the change in log2 MIC appears very high on the y-axis; you can see 4 of these cases for E. coli. The dots with red circles here are R-MIC predictions we think could be influenced by database curation problems because the correlation of MICs between years are not particularly stable, at least not according to our algorithms.
So where does this leave AI, ATLAS and its MICs? Evidently, to make better, safer predictions we need bigger and better MIC datasets, that are, ideally, as free of bias as possible. But getting those data into the hands of quantitative researchers will need investment because it needs hospital labs to pass on all their MICs for curation at all times, not just a select few.
And what can be done thereafter if the necessary infrastructural investments into healthcare data pipelines are made so all that happens? Well, this Lancet Microbe paper is a case in point. It uses raw MIC data from hospitals in Pittsburgh to show that the idea of exploring collateral sensitivity networks between antibiotics to design better multi-drug treatments might not work well in the clinic, even though the idea can work well in the lab, as we have seen ourselves as a research group.
We believe that ruling out novel treatment concepts, and ruling new ones in, is a potential application area of "antimicrobial AI" that we might see in the future, if, that is, national health agencies can work together to publish raw MIC datasets at scale. But until that time comes, we'll have to content ourselves with exploring the potential of AI using ATLAS, despite its variable levels of geographical coverage.
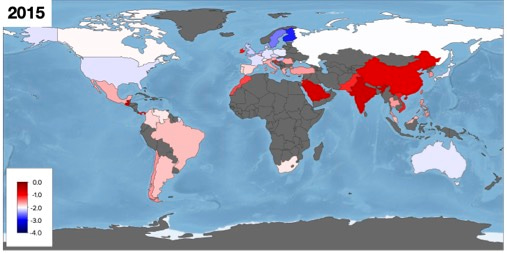
To illustrate the geographical biases, global coverage is shown in the following image where the red-blue colour scale depicts mean log2 MIC levels across all drug-bug combinations (which is not a particularly meaningful concept) and dark grey regions indicate the absence of data:
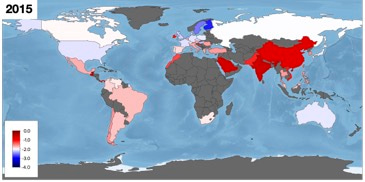
Whatever the future holds for MIC databases, we've learnt that concrete progress can only be made if infection clinicians, with their intimate understanding of antibiotic clinical practise and its history, work very closely with data scientists to prevent AI algorithms from making fanciful predictions.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in