Synthesis of Bridged Tricyclo[5.2.1.01,5]decanes via Nickel-Catalyzed Asymmetric Domino Cyclization of Enynones
Published in Chemistry
![Synthesis of Bridged Tricyclo[5.2.1.01,5]decanes via Nickel-Catalyzed Asymmetric Domino Cyclization of Enynones](https://images.zapnito.com/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95/https://images.zapnito.com/users/386018/posters/1587391955-90-8876/edde0f25-1802-4278-859b-013b1d877537_large.jpeg)
The bridged tricyclo[5.2.1.01,5]decanes are found as core structures in many bioactive natural products, including Schincalide A and Illisimonin A. Despite their importance, the bridged tricyclo[5.2.1.01,5]decane remain an elusive skeleton, and the development of which has been clearly underexploited. The limited output for these challenging molecules may be due to the difficulty in asymmetric synthesis of this bridged tricyclo[5.2.1.01,5]decane core, which hinders any further study on their potential bioactive properties. Therefore, a general approach that enables the modular and enantioselective synthesis of this key skeleton is highly desired.

Inspired by the precedent work on 1,6-enynes cyclization1 and arylative cyclization of alkynones,2 we designed a cyclization reaction of 1,6-enynones for the synthesis of biologically important bridged tricyclo[5.2.1.01,5] decanes via a domino arylnickelation of alkyne/Heck cyclization with alkene/nucleophilic addition to ketone sequence. However, to realize this conceptually simple yet attractive transformation, many challenging problems need to be addressed. The first is to control the regioselectivity of the 1,2-addition of the aryl-nickel species to the alkyne moiety. The second is that ketones are more electrophilic than unactivated alkenes, the direct cyclization of alkynes and ketones may take place preferentially, while unactivated alkenes do not participate in the cyclization process. Another challenge is to control the enantioselectivity of the Heck cyclization process. After a series of conditions optimization, we found that the choice of an electron-rich and conformationally rigid P-stereoisomer bis(phospholane) ligand (1R, 1'R, 2S, 2'S-Duanphos) as the ligand is the key to success.
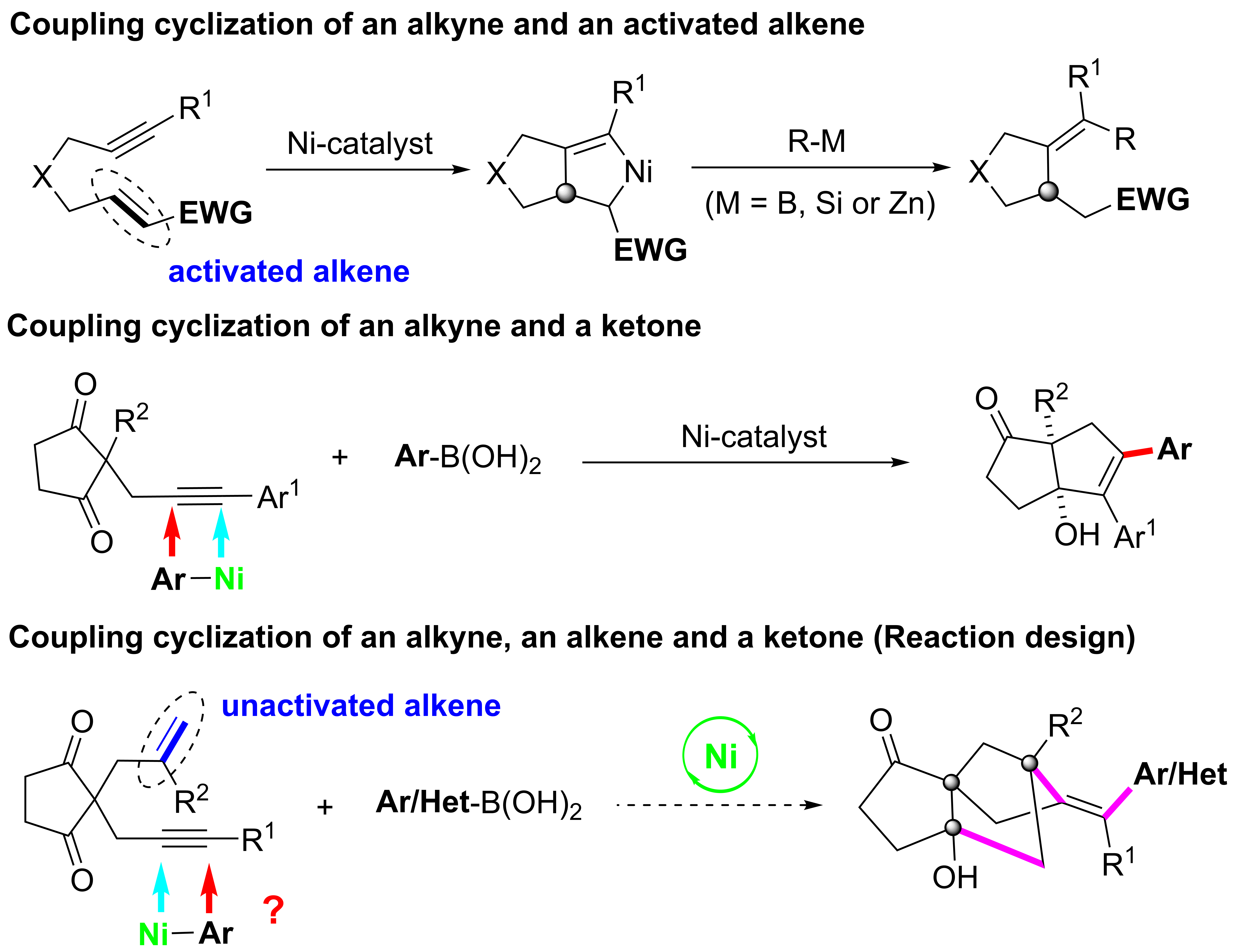
In the present work, a series of bridged tricyclo[5.2.1.01,5]decane skeletons with three quaternary stereocenters were obtained in good yields and remarkable high levels of regio- and enantioselectivities (92-99% ee). The domino strategy not only has the advantage of being efficient, simple and starting from easily accessible precursors, but also provides the desired targets with two additional functional sites (a ketone and a fully substituted double bond), which could be easily used for diversity-oriented synthesis. This work documents a practical, catalytic enantioselective approach to the most diverse set of bridged tricyclo[5.2.1.01,5]decanes reported to-date.
If you want to learn more details about how the bridged tricyclo [5.2.1.01,5]decane is formed, please check our paper here: https://www.nature.com/articles/s41467-020-15837-1
REFERENCES:
[1] Montgomery, J. Nickel-catalyzed cyclizations, couplings, and cycloadditions involving three reactive components. Acc. Chem. Res. 33, 467-473 (2000).
[2] Clarke, C., Incerti-Pradillos, C. A. & Lam, H. W. Enantioselective nickel-catalyzed anti-carbometallative cyclizations of alkynyl electrophiles enabled by reversible alkenylnickel E/Z isomerization. J. Am. Chem. Soc. 138, 8068-8071 (2016).
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in