Targeted nanopore sequencing with Cas9-guided adapter ligation
Published in Bioengineering & Biotechnology
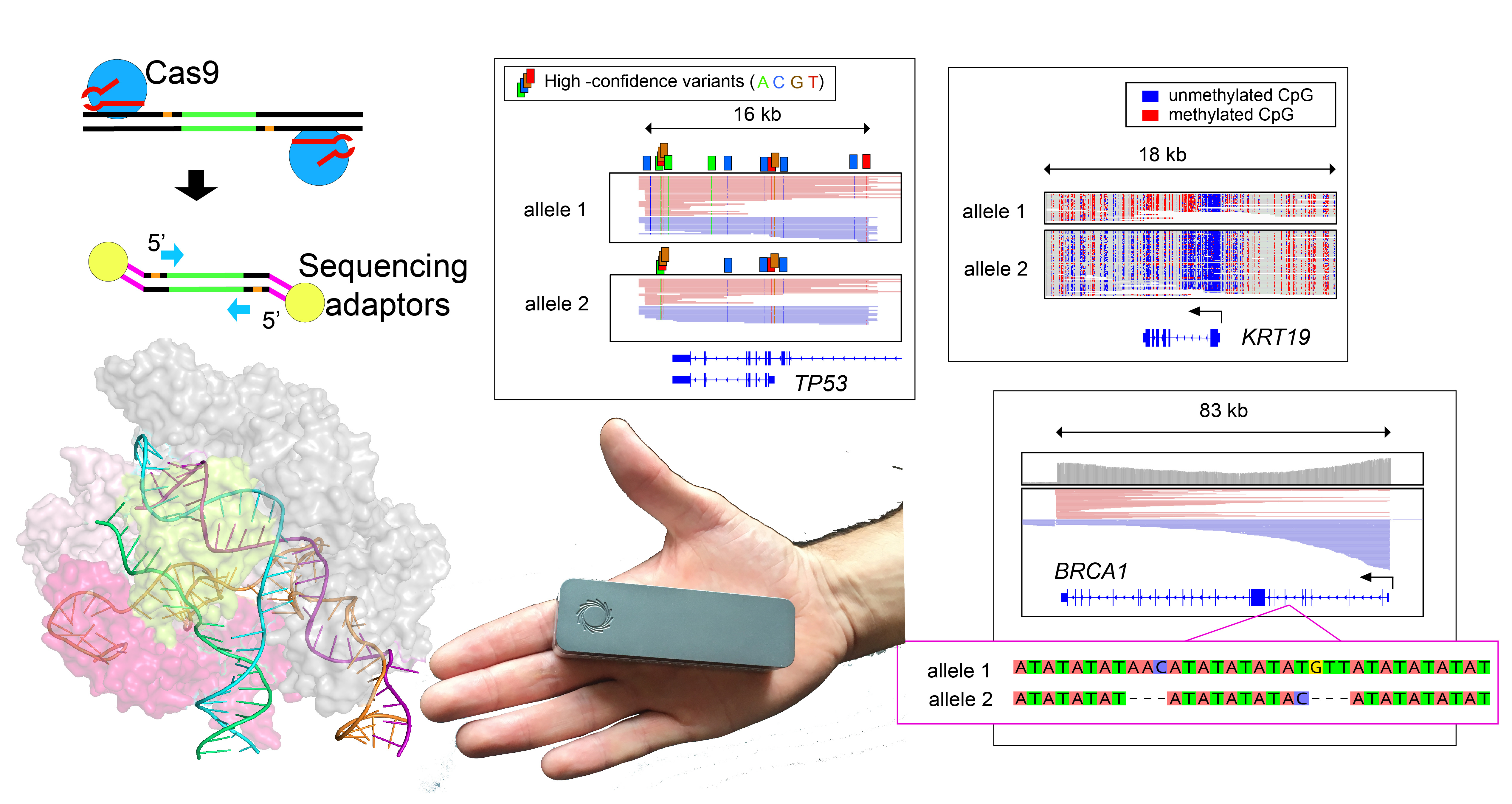
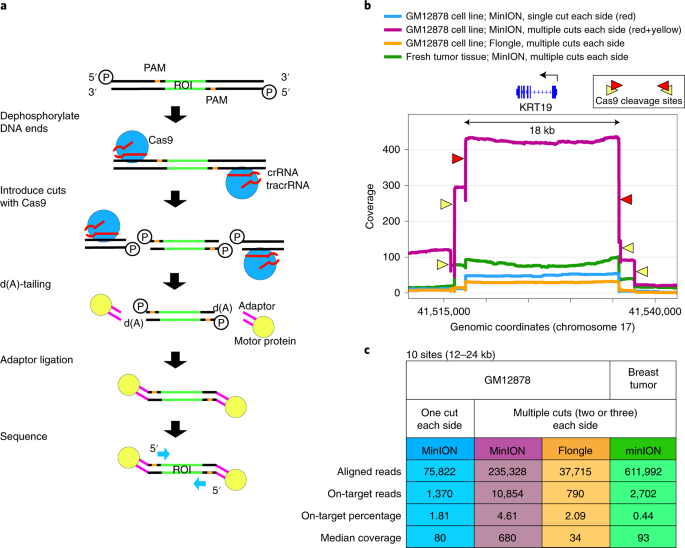
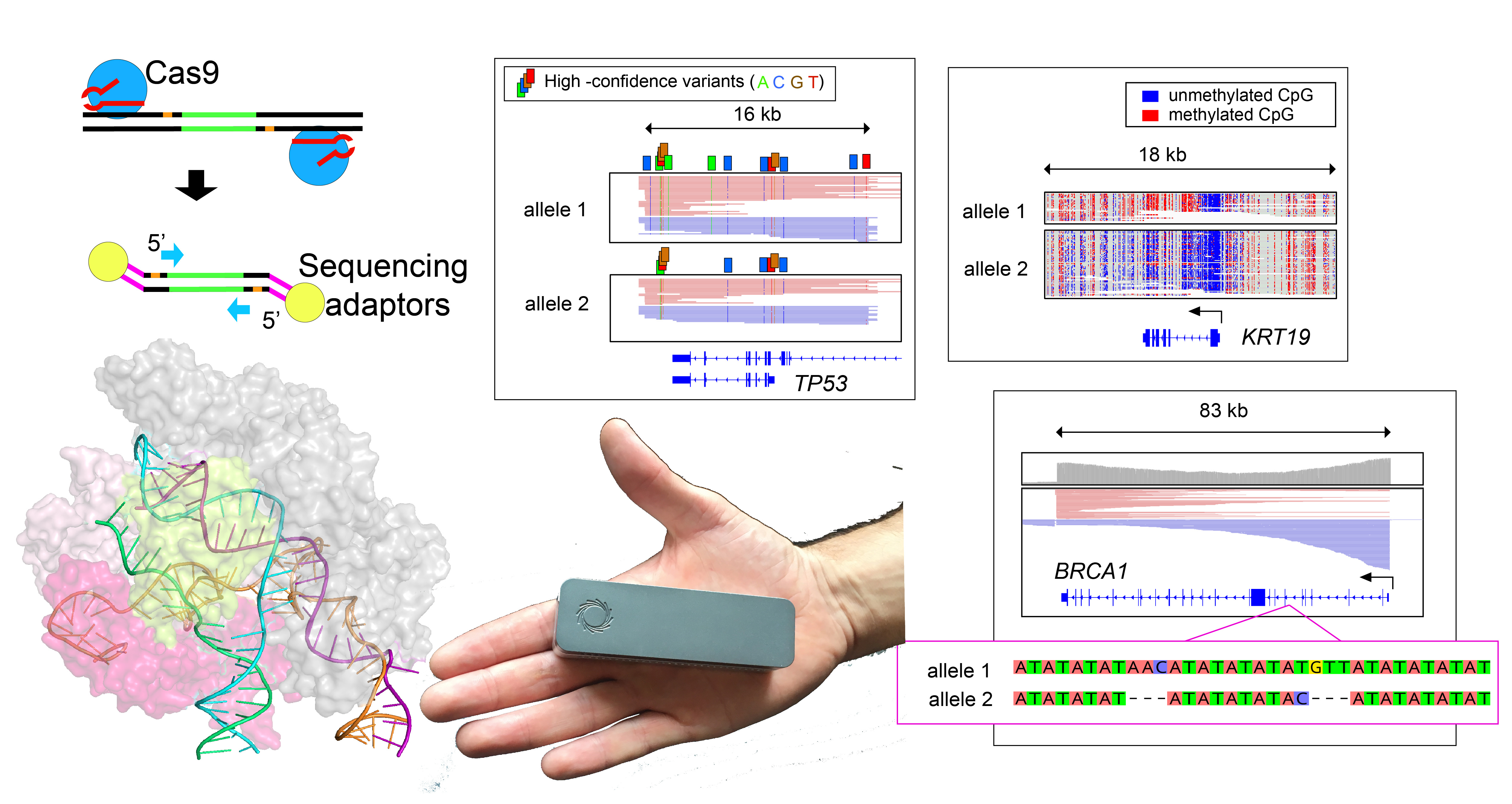
Timothy here --
The work described in our recent Nature Biotechnology publication for Cas9-enrichment with nanopore sequencing began in the winter of 2017/2018 when Oxford Nanopore approached the Timp lab for a collaboration testing and developing strategies for targeted sequencing at select loci. At that time, a passionate and talented colleague of mine, Brittany Avin, was doing work on thyroid cancer, and we thought this would be a good initial avenue to test enrichment methods. Brittany was interested in DNA methylation patterns at the promoter for the TERT gene, which encodes the major protein component of the telomerase complex. It was working with thyroid cell lines at a very early iteration of this assay, when we became excited by preliminary data showing the long reads could discern allele-specific DNA methylation patterns using variants tens of kilobases away from gene promoters. This got us in the Timp lab real jazzed about the potential for this technology to help unveil other nuances of biology, and we began applying this strategy to a breast cancer model. The work in thyroid was taken forward by Brittany and paired with studies of transcription factor binding (that interesting study is under peer review at the time of writing this blog post).
While we continued to experiment with increasing on-target enrichment, simultaneously there were improvements in software tools for calling variants from nanopore data. For instance, while our paper was under development, two neural network based variant callers were released (Clair & Medaka). We found that with high-coverage enrichment data many software tools were able to do an impressive job at recalling single nucleotide variants from noisy nanopore sequencing data. It was spending time emotionally invested in this data and looking for ways to separate ‘true’ variants from ‘false-positives’, that we noted erroneous variant calls by many software tools were the result of errors on only one of the DNA strands (i.e. nanopore signal was consistently miscalling a variant on one direction of the DNA double helix, but the reverse complement strand was read correctly). Surprisingly, to our knowledge, none of the variant calling tools were taking into account this dual-stranded data, and by applying this information we were able to nearly eliminate false-positive variant calls. This allowed us to identify high-confidence variants de novo from nanopore data, which became critical to our studies evaluating a paired tumor/normal sample. Not only were we able to identify tumor-specific variants in cancer driver genes, but also locations of aneuploidy through imbalanced chromosomal copies, along with allele-specific loss of DNA methylation on amplified chromosomes. Being able to assess all of these features helped to uncover insight into this patient's tumor that would be difficult to ascertain by other sequencing strategies.
In addition to methylation and point mutations, we applied the enrichment approach to evaluate structural abnormalities. Initially, this involved looking at candidate DNA deletions identified from whole-genome data and showing that these deletions were robustly confirmed by targeted sequencing. While we were in the early phases of data generation, the Timp lab had the distinct pleasure of attending a talk by visiting lecturer Mary-Claire King, who has spent much of her esteemed career studying the genetics of breast cancer. In her talk, Dr King went into detail about the persisting challenges in resolving variants in the familial breast-cancer-associated genes, BRCA1 and BRCA2. These genes are challenging to sequence because of their large size (>80 kilobases) and because they are highly repetitive (riddled with so-called “Alu-elements”). Although the effects of many mutations in these genes are characterized, there remain regions that are challenging to resolve with existing methods. Accordingly, we elected to test BRCA1 enrichment with our strategy, and were thrilled to find we could generate sequencing reads spanning the entire 83-kilobase gene. In addition, we were able to generate adequate BRCA1 coverage to pass the data to software polishing tools that found numerous small variants, including some previously un-annotated within repetitive regions of BRCA1. This underscored to us that targeted sequencing paired with consensus tools is a valuable strategy for exploring difficult-to-query regions of the genome, providing another tool in scientists’ arsenal for exploring the intricacies therein.
There are a myriad of ways that this strategy can be applied in both the research laboratory as well as in the clinic, and we are excited for all the future iterations and applications of targeted sequencing and the mysteries they help uncover. And just to close out here, without waxing too much emotionally.. I feel really fortunate having been postured to bring this work to fruition and am extremely grateful for everyone and everything that helped bring it together. Thanks ONT folks, bossman, lab mates, and collaborators for all of your support and input and for tolerating me through this.
And thanks to you for reading my blog post-
Yours,
Tim ( gilfunk )
P.S. heres some photos :


Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in