Teaching a machine to read raw spectroscopic data
Published in Chemistry and Computational Sciences
How did the idea emerge?
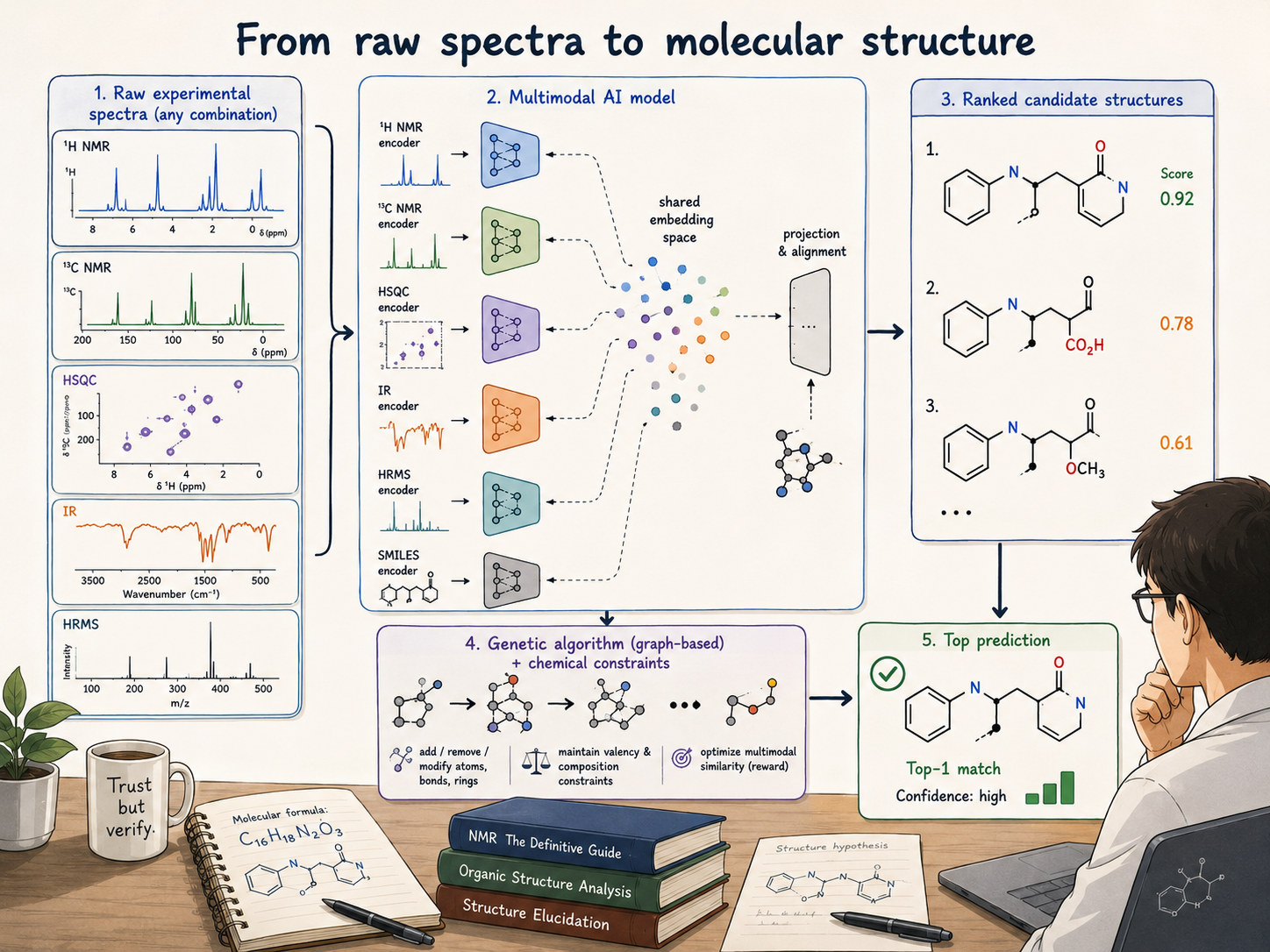
After managing to build a model that aligned different representations of molecules as part of a workshop paper, we started thinking about how such alignment models could be practically useful. Chemists typically interact with real-world molecules not as text representations, but as spectra.
However, at the beginning, the lack of curated experimental data was a big bottleneck , as no such datasets are openly available. So our first experiments were on simulation data in their raw form, not as post-processed peak data. This, in hindsight, was a good choice, because it left open an avenue for directly processing raw experimental spectra. However, the gap between experiments and simulations was still large — that is, if we kept treating the data as-is. In fact, our initial experiments showed no raw spectra elucidation ability. Adding simple artefacts such as noise was also insufficient, even though in this case a first signal gave us hope that the approach would work.
How did we make it work?
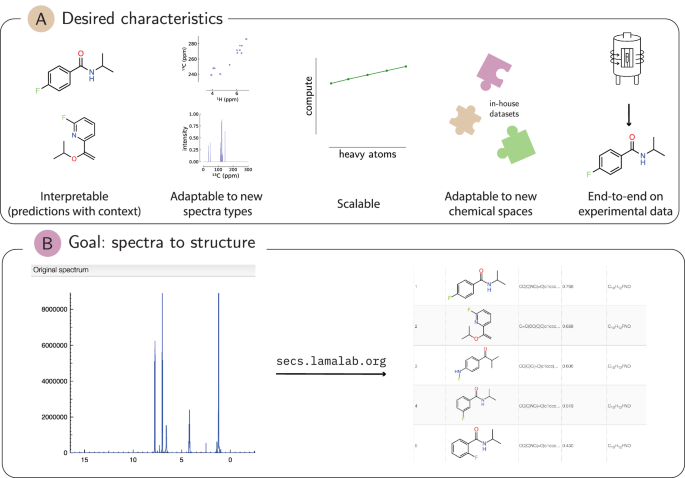
The next months were spent testing different augmentation techniques: essentially trying to make the simulated spectra look more like the experimental ones. A plethora of options were available, ranging from simple noise addition to baseline distortion, peak broadening, and impurity addition. This was promising from the start, and we saw that using simulations alone was sufficient at the initial stage. The real performance upgrade came from using our in-house experimental dataset as a fine-tuning step.
In order to build a better understanding of our pipeline and its results, we worked together with Luc Patiny, who had also built visualization platforms for other Jablonka Lab projects. Together, secs.lamalab.org was built, version by version, with Luc's NMR expertise aiding the project tremendously, as he scrutinised every output of the SECS (Structure Elucidation from Chemical Spectra) pipeline. On a less serious note, there is at least once a week a joke in the lab related to this name.
Initially, the pipeline was served on graphics processing units (GPUs) in the cloud. This was not sustainable in the long run due to prohibitive costs. So we had to find a workaround for our most time-consuming step: converting all available isomers of a molecule into embeddings. The solution turned out to be hiding in vector databases, which can efficiently compress vectors at scale. We took all molecules from the PubChem database as our starting point, from which we retrieve the most similar molecules to the spectrum we want to elucidate. These serve as the starting point in the optimisation process, where we iteratively modify the molecular structure to better match the representation of the spectrum.
How does it compare to chemists?
Thanks to three of my fellow PhD students with chemistry backgrounds and three NMR experts, all of whom accepted a challenge against our system, we managed to obtain a comparison with human chemists. On 20 randomly chosen molecules from our experimental evaluation suite, the system performed competitively — to our surprise even outperforming one of the experts on the chosen spectra.
For autonomous labs to be more than a demo, they need to interpret their own measurements. We don't think SECS is the final answer, but it is an important stepping stone, demonstrating promising results using perhaps the single most used spectroscopic method: proton NMR.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in