The Hidden Flow in Enzyme Catalysis
Published in Cell & Molecular Biology
The Puzzle
Enzyme catalysis, a fascinating biological process, governs numerous essential molecular reactions to maintain cell hemostasis. Therefore, decades of scientific studies have been dedicated to explore and understand how enzymes perform their functions by utilizing a wide spectrum of computational, biochemical and biophysical approaches. In which, determination of enzyme three-dimensional structure provides a direct visual evidence and illustration of how enzymes carry out the mechanistic steps to enable the catalytic reactions. Theories of ligand binding include the earlier lock-and-key, induced-fit, and recent emerging concept of conformational selection. The evolution of these theories is accompanied with advancement of modern technologies in biophysics and engineering. With application of state-of-the-art experimental and computational approaches such as time-resolved x-ray crystallography, cryogenic electron microscopy (cryo-EM), advanced nuclear magnetic resonance and molecular dynamics simulation, structural enzymology has entered a new era. The theories of ligand binding can be further elaborated to provide deeper understanding of how chemistry is synchronized with dynamic features of the protein catalyst.
The Course
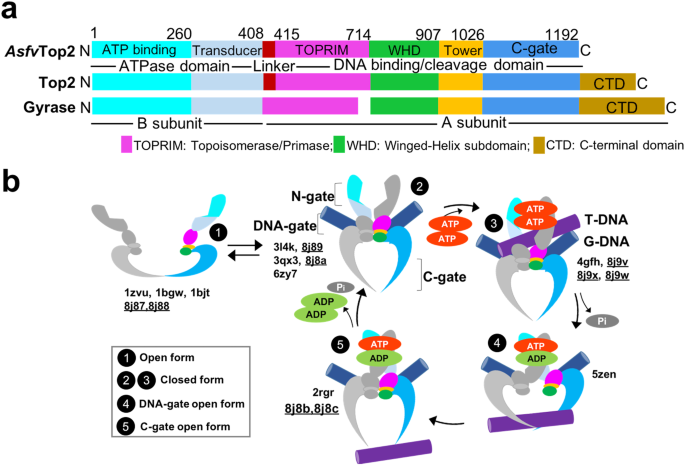
To address the issues above, we used a canonical type II topoisomerase (Top2) encoded by the African swine fever virus (Asfv) genome. Asfv is a large DNA virus that poses a threat to worldwide agriculture. So far there is no effective vaccine or therapeutics. AsfvTop2 has been shown to be involved in the viral genome replication and transcription, albeit its operating mechanism and ligand-bound structure have yet to be characterized. Several decades of biochemical and structural studies of Tops2 from different species have proposed that Top2s change DNA topology by employing a complex ATP-fueled catalytic process, involving well-coordinated changes in protein quaternary structures (Fig. 1). The occurrence of these sequential mechanistic steps is closely related to the dimerization and catalytic activity of the ATPase domain. The binding and sequential hydrolysis of the two ATP molecules allosterically mediate the Top2 core region and thus the steps in its catalytic cycle.

Fig. 1. The proposed catalytic cycle of Top2. The full-process is orchestrated by the binding and release of ligands, including Transport/Gate-DNA and ATP. Together with the sequential ATP hydrolysis, the operating mechanism of N-, DNA, and C-gate is timely regulated. Fig. is adapted from the original publication: DOI 10.1038/s42004-024-01129-y.
We first used cryo-EM to navigate the conformational landscape of the apo-AsfvTop2, which unexpectedly exhibited six conformers, manifesting the dynamic nature of Top2s. These apo-AsfvTop2 conformers resemble the major conformational states of Top2 catalytic cycle (Fig. 1). Conformational conversions amongst these apo-AsfvTop2 conformers are reminiscent of the Top2 gating mechanism (Movie: Conformational conversions of apo-AsfvTop2, adapted from DOI 10.1038/s42004-024-01129-y), suggesting that these motions in the apo state may have been pre-designed for the conformational transitions around the catalytic cycle trajectory, a property of conformational selection in the absence of ligands, which we called “catalytic selection”.
Further, we resolved the cryo-EM structures of the AsfvTop2-DNA-drug complexes to illustrate the conformational transition from the apo-AsfvTop2 to the ligand bound state. The results also demonstrate the conformational selection by ligands (DNA/drug) among the major apo-conformers. Further induced-fit was supported by subdomain reorganization and local conformational changes based on the structural comparison of AsfvTop2-DNA-drug complexes with the apo-conformer in the closed state. Thus, the dynamic trajectory of apo-AsfvTop2 and ligand bound conformers form a sequence with purposely primed conformational changes in the direction towards enzyme catalysis (Fig. 2).
Along the course, we also uncovered structural uniqueness of AsfvTop2-DNA-drug interactions and distinct drug binding modes. These findings provide new insights into Top2 operating mechanisms, and may guide future antivirals development targeting AsfvTop2.
The Branch
The ATPase domain region was not resolved to high resolution in our full-length AsfvTop2 cryo-EM reconstitution, since the domain is loosely connected to the core region by a flexible linker. To provide a comprehensive view of the AsfvTop2 structure, we determined the crystal structure for the ATPase domain separately. During the experiments, we unexpectedly observed an intradomain disulfide bond formation under non-reducing condition. Therefore, we solved the crystal structures of AsfvTop2 ATPase domain in both reduced and oxidized states. Upon oxidation, the ATPase domain structure undergoes significant local conformational changes adjacent to the active site region to enable the disulfide bond formation. Several essential residues are further characterized to show their functional relevance in regulation of the ATP hydrolysis activity and DNA decatenation. This redox mechanism involving disulfide bond formation at the ATPase domain is unique to AsfvTop2, and not to other Top2 family members. The redox regulation of AsfvTop2 ATPase most likely will have similar impact on the catalytic core region of the enzyme. However, the in vivo relevance of this redox mechanism remains to be investigated. Analogous redox regulations were also reported previously for AsfvPolX and AsfvAP. Both are involved in the viral DNA modulation and repair systems. Taken together, it is likely to suggest that Asfv has evolved to adapt the oxidative stress environment by manipulating its own proteins that essential for DNA repair and replication. Further in-depth study is required to explore how this functional regulation of AsfvTop2 affects the gene expression and viral cell cycle, and how this unique functional feature affects AsfvTop2 operating mechanisms. Here we call the conformation-based redox regulation of AsfvTop2 “functional selection” in analogy to the catalytic selection in catalysis.

Fig. 2. The hidden flow in enzyme catalysis. A unified view of the ligand binding theories, which are proposed to operate in a sequential manner: 1. Catalytic (functional) selection, 2. Conformation selection, and 3. Induced-fit, for initiation of catalytic reactions. Fig. is adapted from the original publication: DOI 10.1038/s42004-024-01129-y.
The Flow
In summary, we used cryo-EM, X-ray crystallography and functional analyses to unveil several unique structural and regulatory properties of AsfvTop2. The use of cryo-EM led to the resolution of multiple conformers in equilibrium, an important finding suggesting pre-existence of apo-Top2 in major conformational states of the Top2 catalytic cycle, reflecting the catalytic/functional selection. Then we elucidated the DNA specificity of AsfvTop2, and showed that it binds anti-cancer inhibitors with different sets of active site residues relative to human Top2. By structural comparison, the ligands bind with the most populated apo-conformer, the closed form, followed by local conformational changes (induced-fit). These outcomes lead us to propose that enzymes follow a three-stage mechanism in order (Fig. 2): (1) catalytic selection by the enzyme, (2) conformational selection by the substrates, and (3) induced fit. The flow could be universal in enzyme-substrate or protein-ligand recognitions, despite it is likely unseen. Additionally, several unique structure and regulatory features of the enzyme are unveiled and provide potential implication for antivirals development. The serendipity is hidden behind awaiting exploration.
Follow the Topic
-
Communications Chemistry

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Experimental and computational methodology in structural biology
Publishing Model: Open Access
Deadline: Apr 30, 2026
Advances in Asymmetric Catalysis for Organic Chemistry
Publishing Model: Open Access
Deadline: Mar 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in