The Noise That Wasn't Biology: Cleaning Up Single-Cell Data
Published in Protocols & Methods and Immunology

Single-cell RNA sequencing has transformed how we study the brain and immune system. But like many labs, we kept running into a problem that wasn’t always obvious at first glance: the data looked too noisy, and the noise followed a suspiciously consistent pattern.
It took us a while to realize that the problem was not with our biology but with our sample preparation.
The signal we didn’t ask for
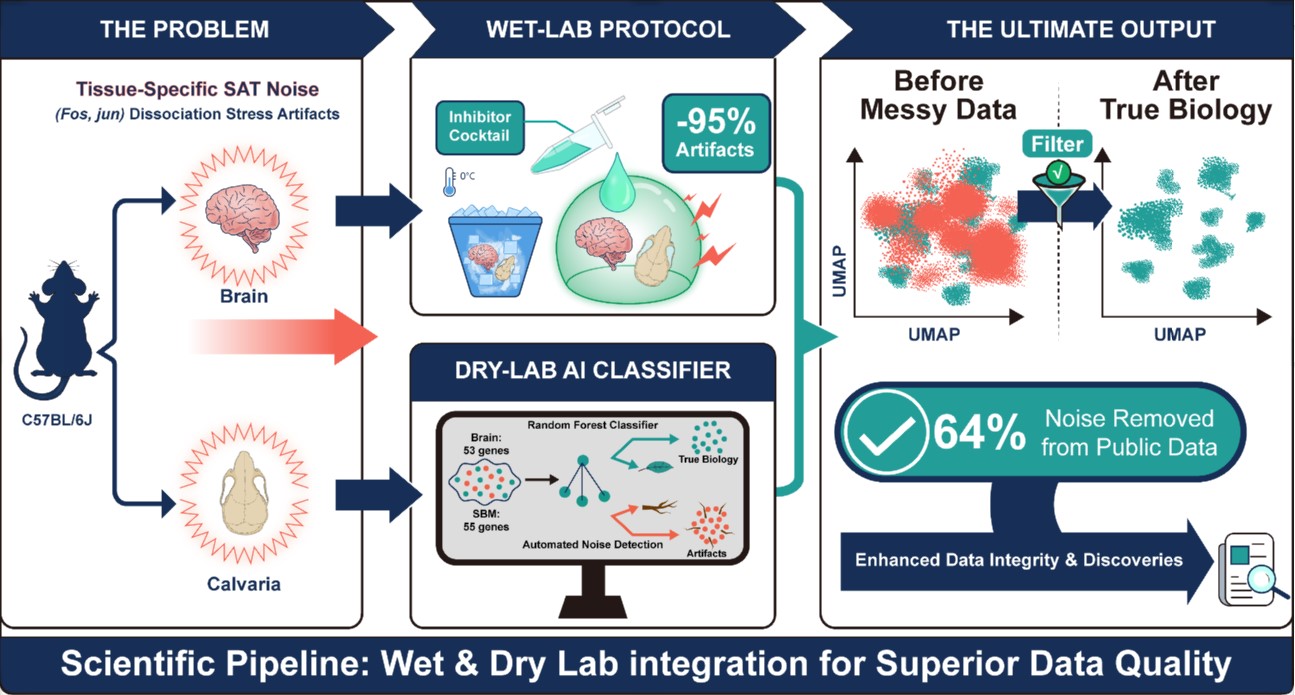
When you break down a solid piece of tissue into a single-cell suspension, you subject the cells to a series of insults: enzymatic digestion, temperature shifts, mechanical forces. The cells respond by mounting a stress response, turning on genes like Fos, Jun, and Hspa1a. These transcriptional changes are perfectly real, yet they are completely unrelated to the biology you are trying to study.
We call this phenomenon “stress-altered transcription” or SAT. And we kept seeing these signatures in nearly every dataset we analyzed. Cells that should have looked “resting” appeared activated. Stress-response genes kept showing up, regardless of the experimental condition. At first, we thought these might be real biological signals. Perhaps we had discovered something new. But the patterns were too consistent, too predictable. And they only appeared in samples that had undergone enzymatic dissociation.
Two problems, two solutions
We realized that solving this problem would require two separate approaches: one at the bench, one on the computer.
On the experimental side, we adapted a cocktail of transcriptional and translational inhibitors consisting of actinomycin D, triptolide, and anisomycin. This combination had been used by others to minimize dissociation stress in the brain. But we wanted to know whether it would work for the skull bone marrow, a tissue where dissociation artifacts had not been profiled. The answer was yes. Our optimized protocol reduced the frequency of artifactually activated cells by about 95%.
But we also knew that not everyone would adopt our protocol. Many labs work with existing datasets generated using standard dissociation methods. We wanted to give them a way to retroactively clean up their data. This is where the machine learning came in.
Teaching a computer to spot artifacts
We trained a random forest classifier to distinguish between cells that were genuinely in a particular biological state and cells that had been pushed into a stress-altered state by the dissociation process.
The challenge was that the signatures of stress and the signatures of genuine biological activation overlap considerably. The same genes, including the immediate early genes, the AP-1 family members, the NF-κB targets, are turned on both by dissociation stress and by real neuroinflammatory processes.
So how do you tell the difference?
We found that while the two states share some core pathways, their overall transcriptional programs are substantially different. The gene expression pattern in a stress-altered cell is not quite the same as that in a genuinely activated cell. With enough data, a machine can learn to distinguish between them.
We identified tissue-specific SAT gene panels: 53 genes for the brain and 55 genes for the skull bone marrow. When we applied this tool to published datasets, we were able to reduce artifact-related transcriptional signals by 60 to 64 percent.
What surprised us
One of the most unexpected findings was that dissociation artifacts are tissue-specific. The SAT signature we identified in the brain looks different from the one in the skull bone marrow. This makes sense in hindsight. Different tissues have different cellular compositions, extracellular matrices, and stress responses. But it also means that researchers working with new tissue types cannot simply adopt our gene panels wholesale.
We also learned that the inhibitors we used are not a perfect solution. They suppress dissociation-induced stress, but they also inhibit transcription and translation more broadly. Any genuine rapid transcriptional response to the tissue microenvironment would also be suppressed. This is a fundamental limitation, and we discuss it candidly in the paper.
Where do we go from here?
Single-cell technologies have given us an unprecedented window into biological complexity. But that window is only as clear as the sample preparation allows. If we are not careful about how we generate our data, we risk building our understanding on artifacts rather than biology.
Our hope is that the framework we have developed will become a standard part of the single-cell workflow, not just in neuroimmunology, but across all fields that rely on dissociated tissue samples.
We are already thinking about the next steps: applying our approach to other tissues, integrating spatial information to better understand where artifacts arise, and refining our computational tools to make them more accessible to the broader research community.
Science is hard enough without having to worry about whether your data are telling you the truth. We hope this work helps researchers worry a little less.
My research focuses on neuroimmune interactions in Alzheimer's disease and brain aging. By integrating multi-omics approaches with integrative bioinformatics and AI, my lab aim to decode the immune mechanisms that regulate neurodegeneration and brain aging, and to develop immune-based therapeutic strategies.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in