![Transforming C–H Bonds on Bicyclo[1.1.1]pentanes](https://images.zapnito.com/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95/https://images.zapnito.com/users/352444/posters/1580946848-64-2574/0d4ca1f5-e451-4579-b458-901fa5c8d9e5_large.png)
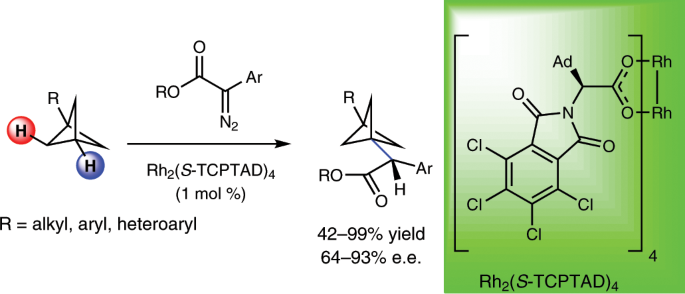
The bicyclo[1.1.1]pentane (BCP) scaffold is a chemical framework which exhibits remarkable topology and considerable strain, and it first garnered the attention of chemists interested in fundamental atomic interactions. More recently, chemists have sought to utilize BCPs as unique building blocks for molecular rods or as a bioisostere in medicinal chemistry. Constructing molecules which possess BCPs is often accomplished by the transformation of [1.1.1]propellane, but this can be undesirable since it has to be made in situ or stored at low temperatures. Instead, what if BCPs that are rich in C–H bonds and stable at room temperature were used as the starting point for generating complexity on BCPs? We considered this question, and our growing interest in this scaffold sparked two further questions. First, we wondered if BCPs could pose a suitable testing ground for selective C–H functionalization through carbene insertion with a dirhodium tetracarboxylate catalyst. Second, if our method was successful and selective, could this new technology be used to construct molecules that are useful to the pharmaceuctical industry?
To achieve the desired goal, the C–H functionalization catalyst would need to discern between secondary and tertiary bonds on the BCP. Our team’s success towards differentiating secondary and tertiary bonds on molecules like adamantane or 2-methylpentane suggested that a positive outcome could also be achieved with BCPs. The BCP scaffold was a more formidable challenge because the C–H bonds appeared to be more inert, and the literature suggested that directly functionalizing this scaffold could be complicated by ring fragmentation and poor selectivity.
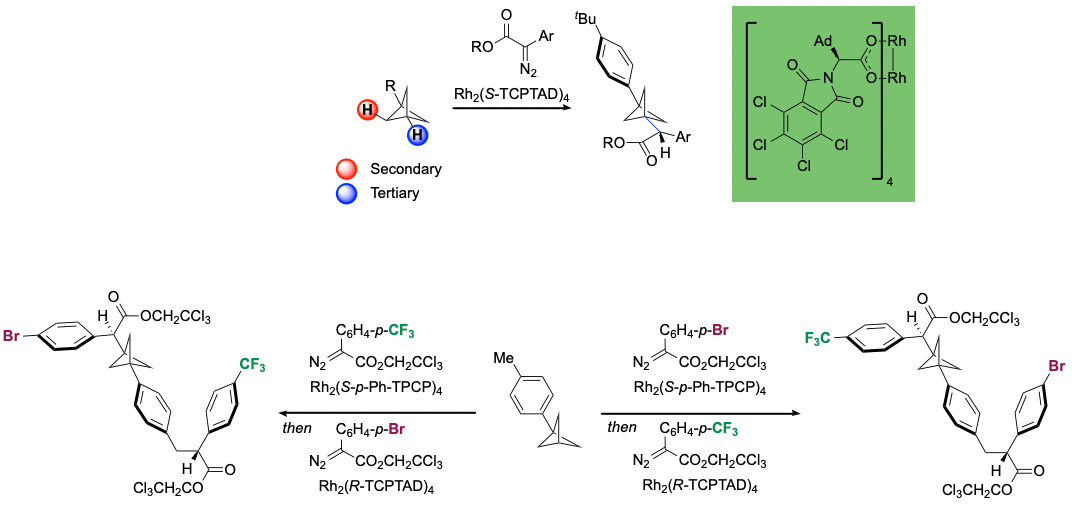
Nonetheless, we tested the C–H functionalization of BCPs using our current suite of catalysts and aryldiazoacetates as donor/acceptor carbene precursors, and we discovered a remarkably selective reaction. Using Rh2(S-TCPTAD)4as catalyst, the C–H functionalization is achieved solely at the tertiary C–H bond with no evidence of functionalization at the secondary C–H bonds or ring fragmentation of the BCP framework. We then used this strategy to build a variety of chiral BCP-containing molecules. Our catalyst suite also permitted sequential selective C–H functionalization reactions wherein a primary benzylic site could be functionalized first and then functionalization of the tertiary C–H bond of the BCP could be achieved on the same molecule. Furthermore, the strategy was used to synthesize a pharmaceutically relevant compound where a BCP ring served as a bioisostere for a phenyl ring. Computational studies unveiled why the transformation occurs only at the tertiary site. During the concerted asynchronous transition state, the developing positive charge which arises at the tertiary site can be sustained across the whole BCP framework.
All in all, we explored our questions and curiosities about BCPs, and we were satisfied by the positive outcome and our newfound ability to construct complex BCP-containing molecules. We hope this report will inspire new imagination towards using BCPs to build more complex molecules with C–H functionalization strategies in mind.
If you are looking for new ways to incorporate BCPs into your compounds or are interested in C–H functionalization in general, check out our article here.
Follow the Topic
-
Nature Catalysis

This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in