Ultra-fast proteomics with Scanning SWATH
Published in Bioengineering & Biotechnology
Background
Our lab has a longstanding interest in understanding cellular metabolism at a system scale. We conducted systematic perturbation experiments to derive the logic of metabolism. For instance, we quantified amino acids in 5,150 yeast knock-out strains that represent the non-essential genome of the biotech workhorse Saccharomyces cerevisiae (Mülleder et al. 2016). In doing so, we learned two lessons: First, the number of genes that are directly or indirectly important for metabolism is huge (⅓ of the genome), despite profiling only one growth condition, and one class of metabolites. Second, we found that metabolic profiles cluster genes according to their function. This approach turned out to be very useful to annotated genes that did not receive a functional annotation by genetic approaches (Mülleder et al. 2016). However, we also learned about the limitations of large-scale metabolomic approaches: The signatures themselves are often difficult to interpret. For instance, why is the deletion of gene W causing a concentration change in the metabolites X, Y and Z? Obviously more information was necessary to answer this question.
To close this information gap, we carried on and started combining metabolomics with proteomics. While metabolomics represented a ‘molecular phenotype’ of the cell, proteomics did reveal changes in the underlying enzymatic pathways (Zelezniak et al, 2018). Having available large and matching datasets, we could go one step further and use machine learning methods. These facilitated the prediction of the metabolome and revealed biological connections and interdependencies between the metabolome and the proteome (Zelezniak et al, 2018). The data-driven approach did reveal multifactorial mechanisms that are difficult to detect in experiments of smaller scale.
Introduction
Our initial combined metabolomic and proteomic study did span over the compendium of yeast kinase knockouts (Zelezniak et al, 2018). Big and systematic enough to make general conclusions about the global nature of kinase functions, but still far away from providing a genome-spanning picture. We realised though that moving to a larger scale was difficult. The available mass spectrometers and software at the time were just not designed for thousands of samples without creating massive costs or creating a lot of noise. We made initial steps to increase the throughput and stability of MS-based proteomics experiments by replacing nanoflow rate chromatography with microflow chromatography (Vowinckel et al 2018), a step that was soon taken also by others (Bian et al. 2021, 2020; Bruderer et al. 2019; Sun et al. 2020). However, the application of short gradients was limited by the speed of the mass spectrometers when operated in data-dependent acquisition (DDA) mode, and the software available for data-independent acquisition (DIA) was not written for the spectral complexity that results from short gradients. To solve the latter, we programmed the DIA-NN software suite (Demichev et al. 2020) that uses deep neural networks to process DIA-MS proteomics data and is specifically designed to support fast chromatographic methods. In parallel, we focussed on developing a semi-automated sample preparation workflow that can process four 96-well plates in parallel and is designed to reduce reagent related batch effects in large studies (Messner et al. 2020); Combining these developments, we converged on running proteomic experiments with 5-minute active chromatographic gradients (800µL/min flow-rate). The increased flow rates allowed us to reduce the overheads to ~3 minutes, allowing 180 measurements per day using SWATH-MS (Gillet et al, 2012). The high chromatographic flow rates also increased chromatographic robustness, resulted in less carry over and resulted in higher spray stabilities (Messner et al. 2020). While the workflow delivered performance suitable for many applications (e.g. COVID-19 biomarker discovery (Messner et al. 2020)) it didn’t reach the proteomic depth and quantitative precision that can be achieved with slower microflow methods. One major bottleneck was the acquisition speed of the mass spectrometer. 5-minute gradient runs with high-flow chromatography result in peaks as narrow as 3 seconds, requiring MS/MS cycle times of 0.5 seconds (see Figure). In conventional SWATH/DIA methods, short duty cycles require wide precursor ion isolation windows, resulting in the co-fragmentation of co-eluting peptides. In addition, short-gradient runs have generally more coeluting peptides due to their lower peak capacities. Therefore, fast proteomic measurements with conventional SWATH/DIA suffered from interferences, limiting signal to noise ratio and reduced quantitative precision.
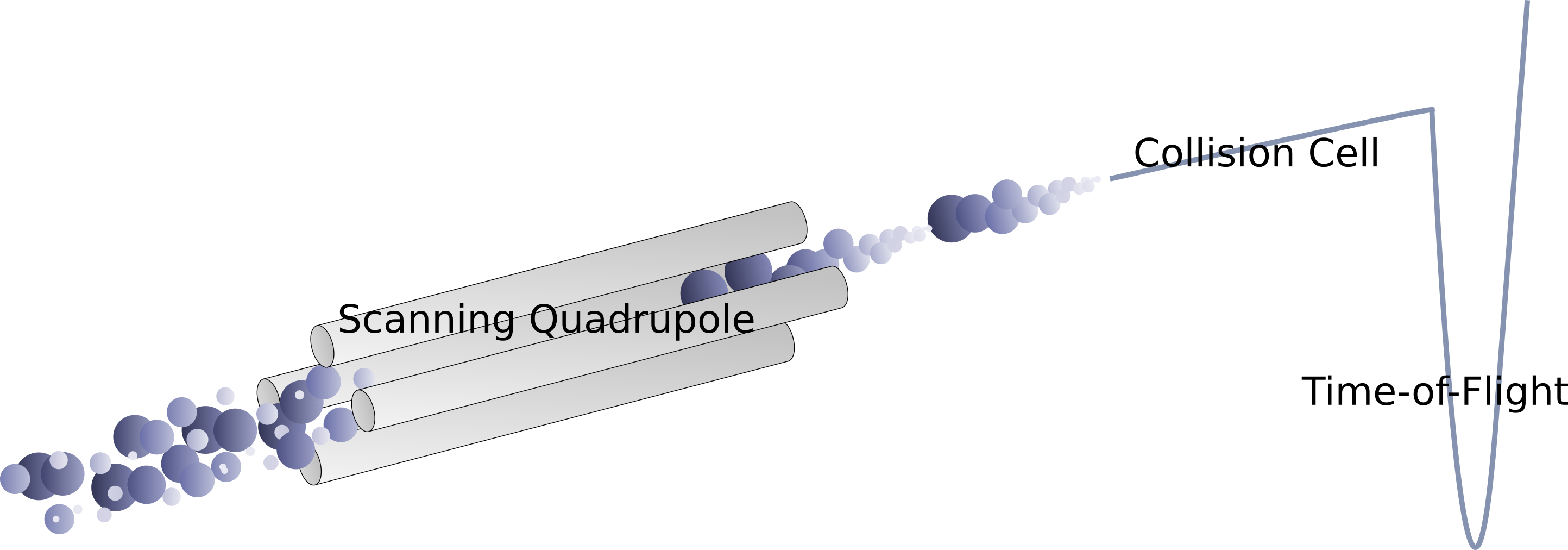
Scanning SWATH implementation
In this situation, we started productive discussions with Stephen Tate and Nic Bloomfield from SCIEX, and during a visit to the Francis Crick Institute we decided to test our fast gradient methods with a new acquisition scheme, which is now known as Scanning SWATH. The method is based on a ‘sliding’ quadrupole (Q1) (see Figure), a concept that was first introduced as part of the SONAR method (Moseley et al. 2018; Juvvadi et al. 2018). We thought a sliding quadrupole could be ideal for high-throughput applications as 1) it would allow shorter duty cycles and 2) adds an additional dimension to DIA-MS data (originating from the time-dependency of fragment signals, when the Q1 quadrupole isolation window slides continuously (see Figure)). At that time we didn't know how we could make use of this additional dimension but we speculated that it could potentially deconvolute the spectral complexity in short-gradient runs.

Figure: Scanning SWATH principle.
During their visit, Steve and Nic did set up a first beta version of Scanning SWATH on one of our instruments. Already after the first tests, we were excited about the combination of short duty cycles and small precursor isolation windows we could achieve by sliding the quadrupole and continuously collecting MS/MS spectra (e.g. 500 ms cycle time/ 5 da isolation windows, compared to 3.2 s/ 25 da in the original SWATH approach (Gillet et al. 2012)). The first version already outperformed our conventional DIA method optimised for fast gradients. To further push the performance and robustness of the technology we spent almost 2 years optimising the algorithms of both the acquisition software and the processing software (DIA-NN). These developments were driven by an extensive exchange and feedback between our lab and SCIEX - in weekly meetings we discussed the latest experiments and implementations.
The key challenge in these developments was to make full use of the additional dimension of information (Q1 dimension) that results from the scanning quadrupole and is unique to this technology (time-dependency of the fragment signals). Nic and Steve implemented an efficient binning algorithm into an Analyst beta version of scanning SWATH. We extensively tested and optimised different parameters and settings and we had to balance file size and information content in the resulting scanning SWATH data files. The final version generates file sizes of 3 GB to 8 GB, depending on the sample type (5-minute runs) and we believe that most labs do have the storage capacities to run such experiments. Further, we tested different Q1 calibration strategies: in the beginning, we calibrated the Q1 profiles “off-line” with direct infusions of a calibration solution but the final version uses an automated algorithm that finds accurate MS1 masses in the data files and matches them to the Q1 profiles. We developed several algorithms that we implemented in the DIA-NN software (15 different scores). These algorithms make use of the Q1 dimension to assign precursor masses to each fragment trace (Figure), which improves confidence in peptide identifications.
New applications are boosted by scanning SWATH and high flowrate chromatography
The throughput of the developed platform allows us to run thousands of precise, low-cost proteomes per week per mass spectrometer. This gives rise to a series of new applications, of which we have presented two examples in the study: we have demonstrated the application of proteomics for antifungal drug screens and COVID-19 biomarker discovery. The former is a new application with huge potential, as proteomes not only capture the phenotypic response of a cell at the molecular level but the mode-of-action. This demonstrates that ultra-high-throughput proteomics can hence combine the advantages of target-based and phenotypic drug screens. In the second example, we have shown that proteomic methods with chromatographic gradients as fast as 60 seconds are sufficient to classify COVID-19 patients according to severity, as well as to identify novel biomarkers.
Ultra-fast proteomes can hence be used as a powerful screening platform in systems biology, drug discovery or medicine. Its low price has the potential to replace less informative readouts in large-scale screens or epidemiological studies. 1-minute gradient runs can potentially acquire more than 100,000 samples per year on one instrument. For example, this would allow the screening of 10,000 different drugs in 3 different cell lines for 3 different concentrations and could be a powerful tool in drug discovery.
Conclusions and Outlook
The applications and benchmarks we present in our study are focused on short gradient runs and complex samples, where Scanning SWATH has the biggest advantage over existing DIA methods. However, we would like to note that the scan mode and the use of the Q1 dimension are also beneficial in longer gradients and with other chromatographic setups.
As instruments will get more sensitive in the future, precursor isolation windows in DIA methods will constantly get narrower and most likely at some point reach unit resolution. In Scanning SWATH methods the window size can be reduced down to unit resolution whereas in conventional stepped DIA methods this is not possible, as overlapping windows would get inefficient and “edge effects” would impact the robustness of the results. We have already demonstrated in the present study that gas-phase fractionation runs with 1 Da scanning windows are a powerful tool for spectral library generation.
Further, the ability to assign precursor masses to fragment traces could be used to improve identifications of post-translational modifications. One such strategy we are exploring in our lab is using Scanning SWATH as a “wide-window” precursor ion scan for oxonium ions to fully map glycosylation states in cells or body fluids.
Finally, we would like to note that setting up a Scanning SWATH experiment is at least as simple as setting up a conventional SWATH method. The only parameters the operator needs to adjust are the precursor range, window size and cycle time. Considering all the advantages that Scanning SWATH has compared to conventional SWATH (the only disadvantage being larger file sizes), we believe that Scanning SWATH could fully replace conventional stepped SWATH on qTOF instruments in the future.
References:
Bian, Yangyang, Florian P. Bayer, Yun-Chien Chang, Chen Meng, Stefanie Hoefer, Nan Deng, Runsheng Zheng, Oleksandr Boychenko, and Bernhard Kuster. 2021. “Robust Microflow LC-MS/MS for Proteome Analysis: 38 000 Runs and Counting.” Analytical Chemistry 93 (8): 3686–90.
Bian, Yangyang, Runsheng Zheng, Florian P. Bayer, Cassandra Wong, Yun-Chien Chang, Chen Meng, Daniel P. Zolg, et al. 2020. “Robust, Reproducible and Quantitative Analysis of Thousands of Proteomes by Micro-Flow LC-MS/MS.” Nature Communications 11 (1): 157.
Bruderer, Roland, Jan Muntel, Sebastian Müller, Oliver M. Bernhardt, Tejas Gandhi, Ornella Cominetti, Charlotte Macron, et al. 2019. “Analysis of 1508 Plasma Samples by Capillary-Flow Data-Independent Acquisition Profiles Proteomics of Weight Loss and Maintenance.” Molecular & Cellular Proteomics: MCP 18 (6): 1242–54.
Demichev, Vadim, Christoph B. Messner, Spyros I. Vernardis, Kathryn S. Lilley, and Markus Ralser. 2020. “DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput.” Nature Methods 17 (1): 41–44.
Gillet, Ludovic C., Pedro Navarro, Stephen Tate, Hannes Röst, Nathalie Selevsek, Lukas Reiter, Ron Bonner, and Ruedi Aebersold. 2012. “Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis.” Molecular & Cellular Proteomics: MCP 11 (6): O111.016717.
Juvvadi, Praveen R., M. Arthur Moseley, Christopher J. Hughes, Erik J. Soderblom, Sarah Lennon, Simon R. Perkins, J. Will Thompson, et al. 2018. “Scanning Quadrupole Data-Independent Acquisition, Part B: Application to the Analysis of the Calcineurin-Interacting Proteins during Treatment of Aspergillus Fumigatus with Azole and Echinocandin Antifungal Drugs.” Journal of Proteome Research. https://doi.org/10.1021/acs.jproteome.7b00499.
Messner, Christoph B., Vadim Demichev, Daniel Wendisch, Laura Michalick, Matthew White, Anja Freiwald, Kathrin Textoris-Taube, et al. 2020. “Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection.” Cell Systems, June. https://doi.org/10.1016/j.cels.2020.05.012.
Moseley, M. Arthur, M. Arthur Moseley, Christopher J. Hughes, Praveen R. Juvvadi, Erik J. Soderblom, Sarah Lennon, Simon R. Perkins, et al. 2018. “Scanning Quadrupole Data-Independent Acquisition, Part A: Qualitative and Quantitative Characterization.” Journal of Proteome Research. https://doi.org/10.1021/acs.jproteome.7b00464.
Mülleder, Michael, Enrica Calvani, Mohammad Tauqeer Alam, Richard Kangda Wang, Florian Eckerstorfer, Aleksej Zelezniak, and Markus Ralser. 2016. “Functional Metabolomics Describes the Yeast Biosynthetic Regulome.” Cell 167 (2): 553–65.e12.
Sun, Rui, Christie Hunter, Chen Chen, Weigang Ge, Nick Morrice, Shuang Liang, Tiansheng Zhu, et al. 2020. “Accelerated Protein Biomarker Discovery from FFPE Tissue Samples Using Single-Shot, Short Gradient Microflow SWATH MS.” Journal of Proteome Research 19 (7): 2732–41.
Zelezniak, A., Vowinckel, J., Capuano, F., Messner, C.B., Demichev, V., Polowsky, N., Mülleder, M., Kamrad, S., Klaus, B., Keller, M.A., et al. (2018). Machine Learning Predicts the Yeast Metabolome from the Quantitative Proteome of Kinase Knockouts. Cell Syst 7, 269–283.e6.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in