Unlocking the Mysteries of 16p11.2 Deletion Syndrome: A Multigenic Perspective
Published in Neuroscience, Genetics & Genomics, and General & Internal Medicine

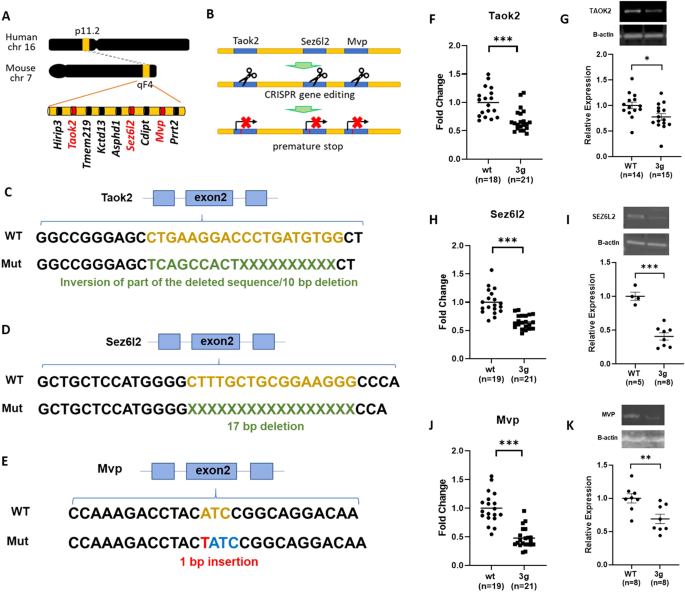
16p11.2 deletion syndrome involves many genes
16p11.2 deletion syndrome is a rare genetic condition caused by a hemi-deletion within the chromosome 16p11.2 locus. This is one of the most common copy number variants (CNVs) associated with neurodevelopmental disorders (NDDs); it is present in 0.6% of individuals with autism spectrum disorder (ASD), and 24% of those with 16p11.2 deletion syndrome also receive an ASD diagnosis. Furthermore, 29% of individuals with 16p11.2 deletion syndrome have attention deficit hyperactivity disorder (ADHD), and 30% have intellectual disability.
CNVs with high effect sizes for NDDs involve multiple genes, and they offer a valuable avenue for studying the polygenic nature of NDDs. Variation in the 16p11.2 region is particularly useful due to its high conservation in the mouse genome, allowing scientists to effectively model this CNV in mice. The region encompasses more than 25 genes (approximately 25-30), providing a unique opportunity to explore how these multiple genes interact, contributing to the increased risks of various NDD phenotypes. However, pinpointing the critical genes responsible for NDD phenotypes within a CNV remains a major challenge in the field, and despite these advantages, it remains unclear which specific genes within the 16p11.2 region contribute to NDD-relevant phenotypes.
Seeking candidate genes
Studying a single gene provides a straightforward, although not entirely sufficient, method to understand the role of a candidate gene within a CNV. Several studies have investigated individual genes within the 16p11.2 region, replicating certain 16p11.2 deletion phenotypes. However, these single gene approaches are unable to examine the interacting effects of multiple genes within the 16p11.2 region.
Rather than focusing on individual genes, we decided to investigate a set of candidate genes within 16p11.2. The challenge we faced was selecting a subset of candidate genes from a huge pool of possibilities. For instance, choosing 2 genes from the 27 within the 16p11.2 region results in 702 potential combinations, and picking 3 genes leads to overwhelming 17,550 possible combinations. We needed a rationale strategy to select candidate genes.
To overcome this hurdle, we turned to our prior work and an innovative approach to identify candidate genes based on their spatial gene expression patterns within structurally altered brain regions in 16p11.2 deletion mice. Utilizing magnetic resonance imaging (MRI) and the Allen Mouse Brain Atlas gene expression map, we pinpointed a set of candidate genes linked to sex-specific structural changes in the striatum of mice modeling 16p11.2 deletion. The identified genes were thousand and one amino acid protein kinase 2 (Taok2), seizure-related 6 homolog-like 2 (Sez6l2), and major vault protein (Mvp).
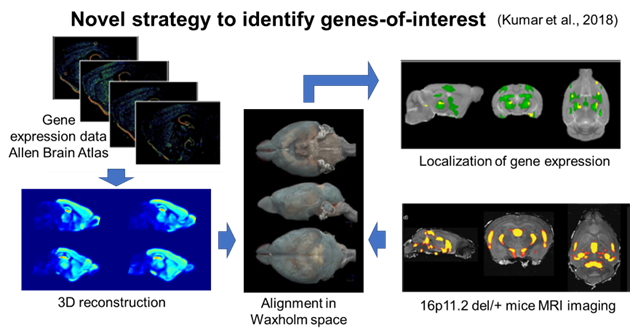
Generating a novel mouse model
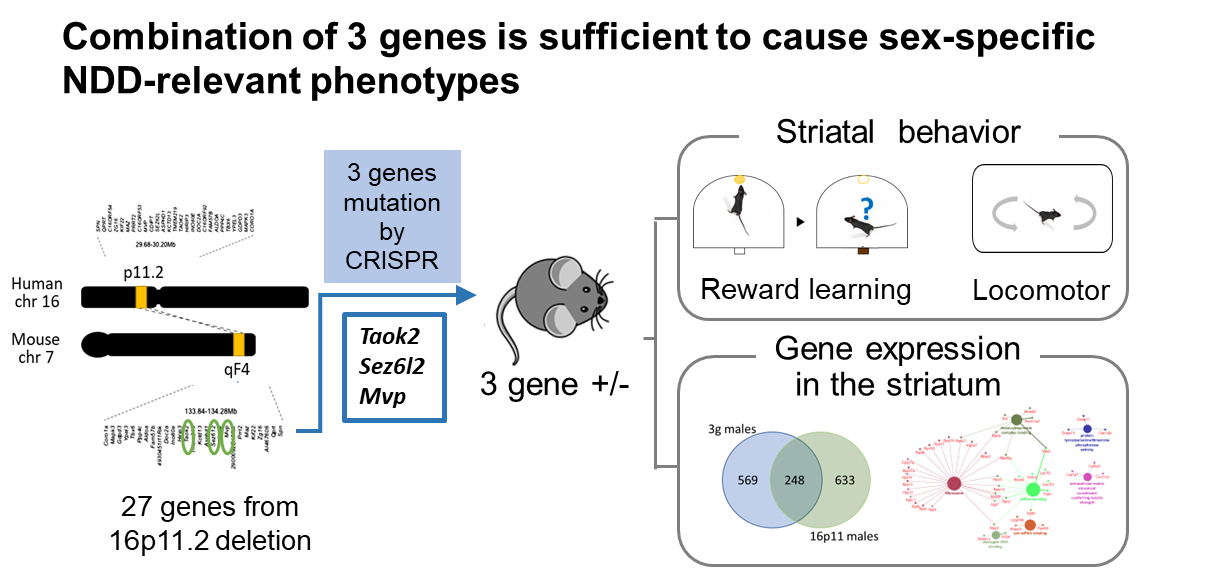
We generated a mouse model with truncating/null mutations in Taok2, Sez6l2, and Mvp. Generating mice with mutations in all three genes by crossing existing single gene knockout (KO) mice proved unfeasible due to the close proximity of these genes in the genome. To overcome this challenge, we employed the CRISPR/Cas9 system to develop a 3 gene hemi-deletion mouse (3g mice), a novel model allowing us to explore the combined effects of our 3 candidate genes on sex-specific striatal behavioral phenotypes and transcriptomic alterations.
What we found
3 gene hemideletion mice phenocopy sex-specific NDD-relevant phenotypes
We observed that the 3g mouse model reproduced key behavioral and transcriptomic changes seen in16p11.2 del/+ male mice. These changes included hyperactive behavior and a reduced motivation to work for rewards, behavioral domains that also show changes in actual patients with NDDs. RNA-seq analysis of the striatum indicated that the 3g mouse model reproduces sex-specific transcriptome changes seen in 16p11.2 deletion mice. These findings strongly suggest that disruption of a small set of genes within a large copy number variant (CNV) is adequate to induce sex-specific phenotypes. In-depth transcriptome pathway analysis points to ribosomal dysregulation as a molecular mechanism contributing to the sex-specific, striatum-dependent behavioral alterations characteristic of NDDs.
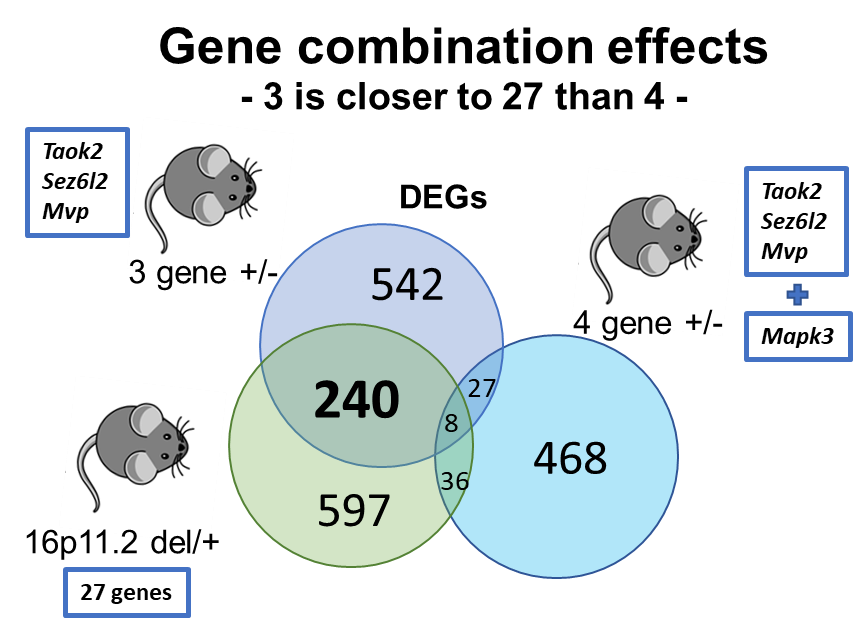
Gene combination effects are not simply additive
We also investigated the role of Mapk3, a fourth gene within the 16p11.2 region, in male-specific phenotypes alongside the three identified genes. ERK1 signaling, encoded by the Mapk3 gene, has been suggested to underlie phenotypes observed in 16p11.2 del/+ mice and has been the focus of potential therapeutic approaches. Our expectation was that mice with a hemi-deletion of four genes, including Mapk3, would exhibit even more pronounced phenotypic similarities with 16p11.2 deletion mice compared to 3g mice. Surprisingly, the results demonstrated the opposite. Mice with the deletion of four genes displayed fewer phenotypic similarities with 16p11.2 deletion mice. This unexpected outcome challenges the notion that the consequences of gene deletions are simply additive, highlighting a crucial distinction between the 'combinatorial effects of genes' and the 'sum of single gene effects.' It emphasizes the need for a better understanding of how many individual genetic influences interact to contribute to the observed phenotypic outcomes in NDDs.
Conclusions
This study underscores the validity of our innovative approach to gene selection, which relies on the spatial gene expression patterns in structurally altered brain regions, highlighting the significance of a systematic and spatially informed approach in gene selection for further investigations. Furthermore, our work emphasizes the importance of adopting a multi-/polygenic perspective when studying NDDs. By exploring the combined effects of multiple genes, we gained valuable insights into the intricate interplay that leads to transcriptomic changes in the brain, eventually resulting in behavioral alterations. The insights gained from this study have the potential to pave the way for novel therapeutic and preventive strategies for NDDs. This strategy could be expanded to incorporate studies of humans with NDDs. Combining human gene expression data with polygenic scores and neuroanatomical changes associated with NDDs will inform the generation of valid animal models that promise to uncover the cellular and molecular mechanisms mediating NDD-relevant behavior resulting from complex polygenic influences.
Follow the Topic
-
Molecular Psychiatry

This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.
Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcement

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in