What brown algae taught me about the importance of jumping genes for genome evolution
Published in Ecology & Evolution and Genetics & Genomics
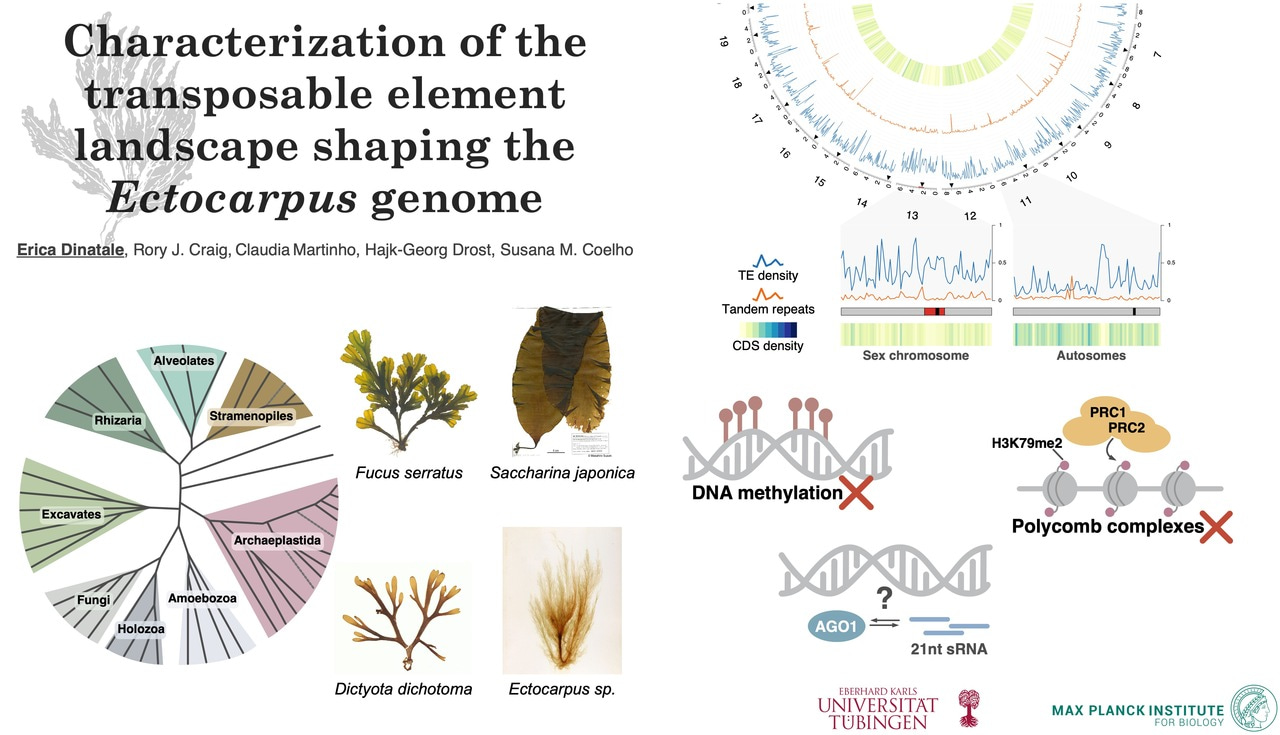
Brown algae represent one of the most complex eukaryotic lineages, but share no recent evolutionary history with animals, plants, or fungi. They evolved multicellularity and embryo development independently, and display remarkable diversity in their developmental programs and sex-determination systems. In that sense, they act as a “biological replicate” in the tree of life. When biological replicates are similar, we usually assume that a particular biologically phenomenon can be reproducibly captured, whereas when they differ, we study more closely where this variance comes from. Analogously, when we find the same patterns or mechanisms across the tree of life (similar replicates), we can assume that underlying mechanisms may also be conserved, whereas when we find differences across the tree of life (variable replicates), we can study more closely what the causes for these differences may be.
One such pattern is how organisms across eukaryotic lineages coexist with transposable elements (hereafter TEs), mobile DNA sequences that increase their copy number by moving within the host genome. In most plants and animals, TEs are kept in check through DNA methylation, chromatin modifications, and small RNAs (hereafter sRNAs). Brown algae, however, show negligible levels of DNA methylation and lack the canonical histone modifications usually involved in TE silencing. So how do they keep these mobile sequences under control?
That question became the foundation of my recent publication, though it all started with much more practical goals.
Why study transposable elements?
The story of TEs began in the 1940s, when Barbara McClintock discovered that certain DNA sequences could move from one chromosomal position to another, disrupting gene function and producing variegated maize kernels. When she presented her results at the 1951 Cold Spring Harbor Symposium, her peers were skeptical, dismissing the phenomenon as a maize-specific oddity and ignoring its functional implications. Yet McClintock was convinced that the phenomenon was universal and that she was witnessing the genome reshaping itself in real time.
For decades afterward, TEs were considered abundant, but functionally irrelevant, selfish passengers in the genome. With the rise of genomics, however, their influence became undeniable. In fact, TEs are ubiquitous across the tree of life and interestingly show highly diverse proportions across even closely related species. They also influence gene expression and phenotypic variation, drive genome plasticity, and allow organisms to rewire their genomes under stress. McClintock’s discovery revealed the dynamic nature of genomes constantly “negotiating” with its own mobile elements when mobilisation can be tolerated or when it cannot.
Despite the increasing number of organisms studied and biological questions answered in the context of TE research, their importance within brown algae has been overlooked: does the remarkable diversity within brown algae extend to their TEs, and might TEs contribute to that diversity?
Transposons research comes with challenges…
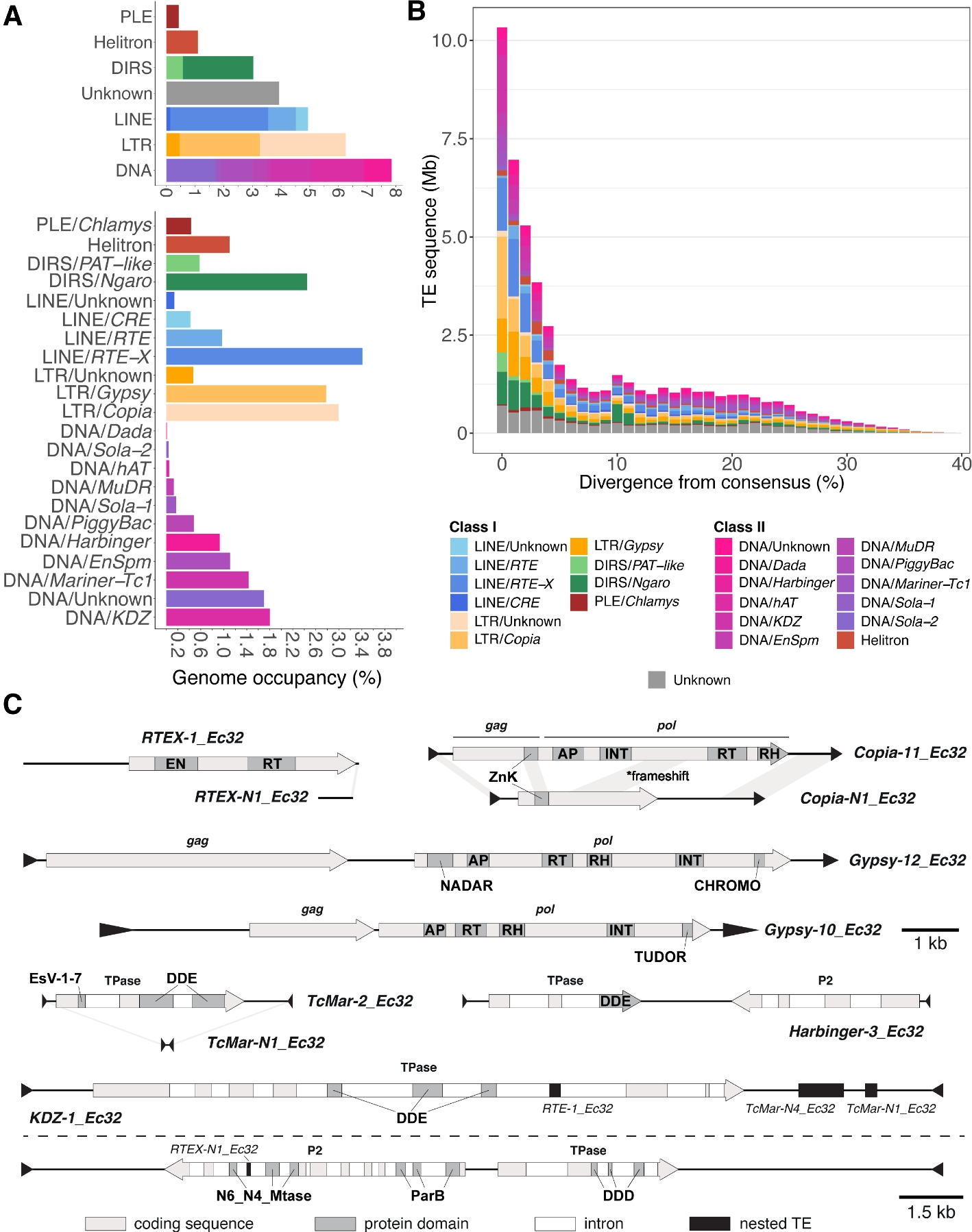
This project began with a concrete task: to build a high-quality TE library for the model brown alga Ectocarpus and make it publicly available as a community resource. A previous fragmented library existed, but the newly improved genome assembly allowed us to examine TEs at a resolution never possible before.
We first applied automated annotation tools to multiple brown algal genomes, resulting in a first general picture of TE diversity and abundance in the group. However, automated tools are typically optimised for plants or animals, leaving many TE sequences unclassified. To move further, we turned to manual curation and reduced the number of unclassified elements while assigning proper classification where needed. This process requires patience and deep knowledge of TE biology, but allows us to reveal unexpected diversity. For those who wish to embark on this journey themselves, I suggest an accessible guide.
Once the library was complete, the next challenge was to disentangle the repetitive nature of TEs from our results. In fact, mapping reads onto TEs could inflate the results, if all reads aligning to these repetitive elements were counted equally. The key here was to allow reads that match multiple genomic positions to align only where they fit best, or to one randomly chosen position only. This way, we can preserve the complexity of TE biology without being distracted by signals hard to dissect.
…and brings new perspectives
With this foundation, Ectocarpus began to reveal its TE secrets.
- The Ectocarpus genome harbors a highly diverse portfolio of evolutionarily young TEs. DNA transposons, which replicate independently of RNA, are predominant over retrotransposons.
- The pseudo-autosomal region (PAR) of the sex chromosomes shows a TE enrichment compared to autosomes, despite similar recombination rates. We hypothesize that it might arise from “hopping” of DNA transposons from the non-recombining sex-determining region (SDR) of the sex chromosomes into nearby regions of the same chromosome.
- Over 80% of genes contain at least one intronic TE insertion. These intronic elements are shorter and more degraded than intergenic TEs, and show minimal impact on gene expression, suggesting ongoing selection.
- Young, potentially active TEs are preferentially associated with small RNAs and with the unconventional histone modification H3K79me2. The two epigenetic mechanisms co-exist in a large proportion of organisms, suggesting an interplay between the two TE silencing strategies.
- The association between TEs and sRNA remains mostly stable across the Ectocarpus life cycle, although in gametes the number of TEs with differential sRNA accumulation is higher, potentially reflecting changes in TE activity during germline formation.
Take home message
Working on Ectocarpus has been a reminder that biology is far more inventive than our standard models suggest. Brown algae lack DNA methylation and the canonical histone marks used by plants and animals to silence TEs. Yet, they have evolved an efficient system relying on unexpected combinations of chromatin modification and sRNA.
The dynamic changes in TE-sRNA associations during gametogenesis suggest that transposon regulation may be intertwined with developmental transitions, as observed in other species. This resonates with a recent publication, first-authored by my friend and PhD collegue Sodai Lotharukpong which identified brown algae as a promising model for studying the evolution of multicellularity and the emergence of embryogenesis as a constrained developmental process shaping body plans. Could there be causal links between TE regulation and developmental innovation?
Brown algae show us that there are many ways to build complexity and regulate genomes, and that some original evolutionary solutions may be hidden in the non-model organisms we have yet to fully explore.
Erica is a doctoral researcher in the department of Algal Development and Evolution. She is currently working on transposable element biology in brown algae 🍙
Follow the Topic
-
Genome Biology

This journal publishes outstanding research in all areas of biology and biomedicine studied from a genomic and post-genomic perspective.
Related Collections
With Collections, you can get published faster and increase your visibility.
Tackling large-scale genomic studies
Genome Biology is calling for submissions to our Collection on large-scale genomic studies. Modern genomic research now generates datasets of unprecedented scale spanning population cohorts, large single‑cell atlases, and high‑throughput multi‑omics studies. These expansive datasets offer powerful opportunities to uncover biological mechanisms, but they also introduce major challenges in data management and interpretation. Addressing these demands requires scalable approaches capable of extracting meaningful insight from large and heterogeneous genomic resources.
This Collection invites contributions that advance the design, execution, and interpretation of large‑scale genomic studies, including:
- Experimental and sequencing strategies optimised for high‑throughput, population‑scale data generation
- Frameworks for data harmonisation and standardisation, enabling cross‑study comparability, meta‑analysis, and integration of datasets generated across platforms, cohorts, or populations
- Scalable machine learning and AI approaches designed for high‑throughput genomic data
- New research with biological insights derived from large‑scale genomic analyses
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Dec 05, 2026
Improving the gene editing toolbox
Genome Biology is calling for submissions to our Collection on recent progress in genome editing technology developments.
Such developments offer exciting opportunities to reshape our approach to understanding biology, driving progress in various fields such as gene therapy, medicine, and agriculture. For example, technologies including base editing or prime editing enable users to install targeted gene modifications via single base substitutions or nearly any short sequence edits, respectively, CRISPRa and CRISPRi can regulate gene expression, various emerging technologies are being explored for exon- or kilobase-scale sequence edits, and the use of AAVs offers potential for studying effects of perturbations in vivo in mammalians. As computational approaches improve, larger-scale CRISPR screens paired with high-dimensional read-outs present the potential to map gene interactions with enhanced accuracy. These breakthroughs are reshaping biotechnology, driving progress in medicine, agriculture, and synthetic biology.
This Collection seeks to highlight research aimed at capitalizing on these innovations across various fields, from fundamental research to therapeutic applications and crop improvement.
We welcome contributions that explore:
Exploiting alternative endonucleases to improve efficacy or specificity
New approaches for exon- and kilobase-sized genome edits
Computational methods/models for rational or de novo design of genome editing reagents or prediction of genome editing efficacy
Efficient delivery systems of genome editing reagents in plants and mammals
Large-scale CRISPR-based screens, including effects of combinatorial perturbations and gene function discovery
Single-cell and other high-dimensional readouts, including Perturb-seq, CROP-seq, ATAC-seq, and optical pooled screens
Mechanistic and structural insights into genome editing tools to elucidate their modes of action and guide further optimization
Engineering programmable gene circuits and synthetic regulatory systems using genome editing platforms
Development of highly specific, efficient, and durable tools for targeted epigenomic editing
Expanding the scope and applications of RNA-targeted editing technologies
We also welcome submissions on any other innovative research that contributes to the advancement of genome editing technologies, even if not explicitly listed above. Authors are encouraged to contact the editors for pre-submission inquiries regarding topic suitability.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Jun 23, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in