A Chiral Bifunctional Superbase Catalyst Forges P(V) Stereocenters by Nucleophilic Substitution
Published in Chemistry
Compounds containing one or more phosphorous atoms in the P(V) oxidation state are important to chemistry, biology and medicine (Figure 1).[i] These include marketed antiviral drugs such as such as Tenofovir alafenamide and Remdesivir, the latter an effective treatment for Ebola[ii] which has currently also been approved for use against SARS-CoV-2.[iii] Other relevant compounds include WHO essential medicines listed chemotherapy agent Cyclophosphamide.[iv] In addition to being common APIs, P(V) containing compounds are also highly relevant motifs in agrochemistry. A representative example is Dow herbicide Zytron.
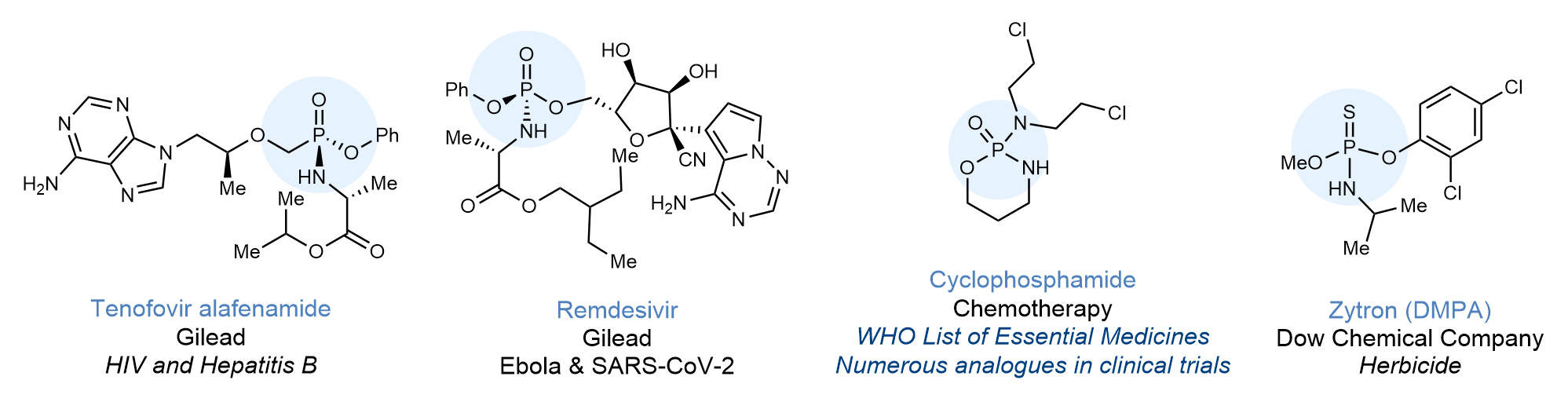
Figure 1. Representative bioactive compounds bearing a phosphorous (V) stereocenter.
Synthetic approaches to enantiopure P(V) compounds can be subdivided into five categories (Figure 2).[v] The first approach is classical resolution and/or separation of diastereomers or enantiomers by chromatography (Figure 2, i). A second approach revolves around the use of labile chiral auxiliaries which can be displaced by sequential diastereoselective nucleophilic additions to afford enantioenriched P(V) species (Figure 2, ii). Thirdly, a racemic and electrophilic P(V) compound bearing a single leaving group (and often possessing a chiral sidechain) can be coupled diastereoselectively (and catalytically) to a chiral nucleophile (Figure 2, iii). A subsequent approach involves the direct functionalisation of secondary phosphine oxides (SPO) by means of a metal catalyst bearing chiral ligands to obtain tertiary phosphine oxides. To date, however this approach has been widely limited to the synthesis of all carbon substituted phosphine oxides (Figure 2, iv). The final strategy is enantioselective desymmetrisation by which prochiral groups attached to a phosphorous centre are differentiated by a chiral catalyst (Figure 2, v). Multiple reactions have been developed using this strategy, however, at the inception of this project, none involved chemistry occurring at the P atom directly but rather generate the P stereocenter indirectly (via manipulation of enantiotopic side chains) inherently limiting their scope.
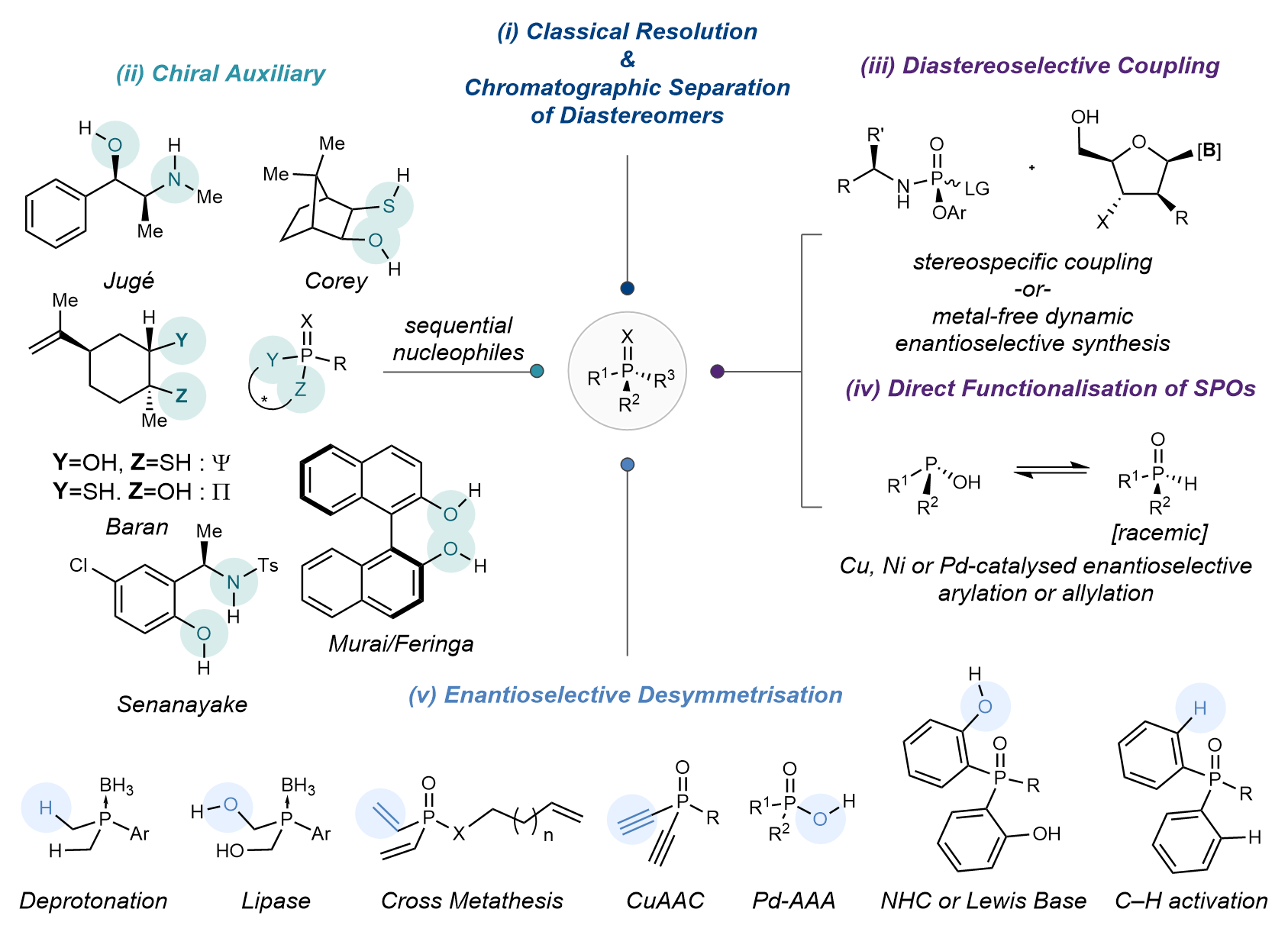
Figure 2. Classical and contemporary approaches to the stereoselective synthesis of enantioenriched, stereogenic at P(V) compounds.
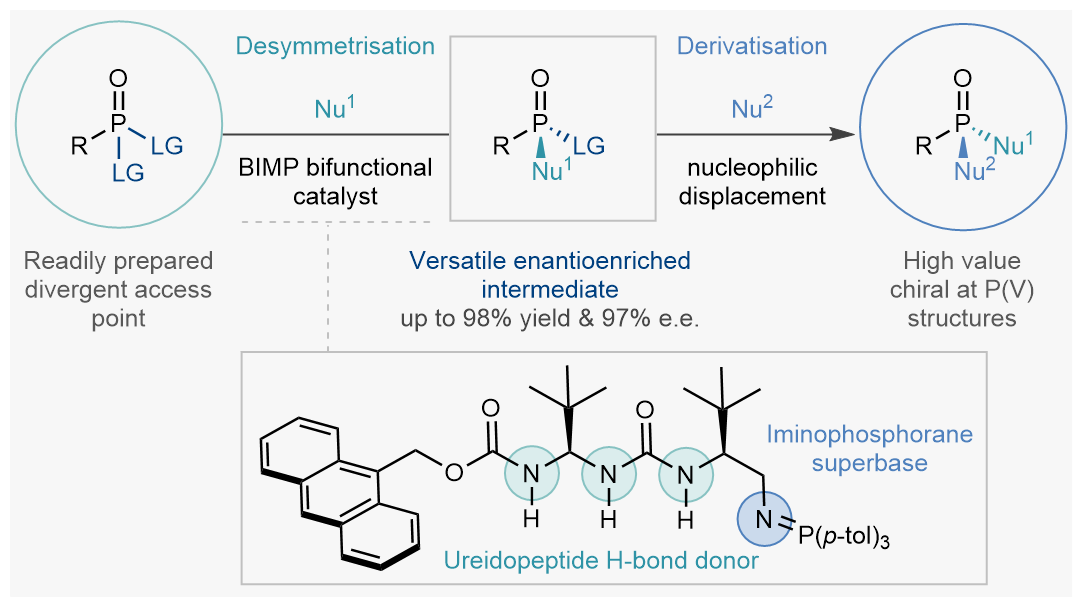
We envisioned a strategically distinct enantioselective desymmetrisation of P(V) species by enantioselective nucleophilic substitution of enantiotopic leaving groups. By judicious choice of leaving group, nucleophile, and catalyst we could obtain the desired enantioenriched species in high yield and with excellent enantioselectivity (phase 1). The desymmetrised substrate would then still possess another leaving group, perfectly poised for a second (potentially enantiospecific) nucleophilic substitution reaction enabling diverse downstream derivatisation opportunities (phase 2). This two-phase strategy would allow for a wide range of P(V) compounds to be accessed from a common intermediate, overcoming one of the main limitations of current enantioselective desymmetrisation approaches (Figure 3).[vi]
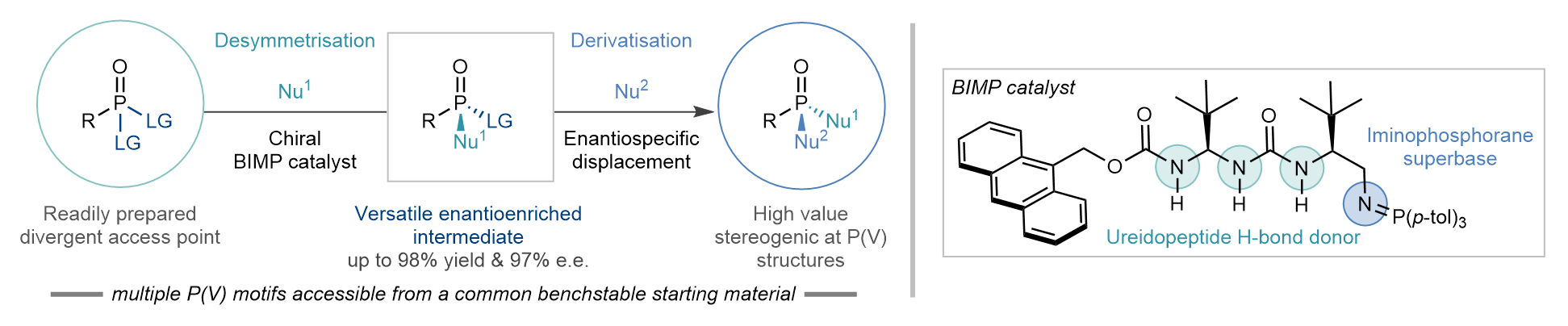
Figure 3. This work: A conceptually different strategy consisting of two separate phases (1) desymmetrisation and (2) derivatisation. In the desymmetrisation phase a ureidopeptide derived BIMP catalyst can effect an enantioselective nucleophilic discriminating between two identical leaving groups on readily accessible prochiral P(V) compounds. In the derivatisation phase the second leaving group can be replaced enantiospecifically by a variety of N-, O- and S- based nucleophiles providing access to a wide range of enantioenriched stereogenic at P(V) architectures from a small collection of enantioenriched intermediates.
When using ortho-substituted phenols as nucleophiles, a ureidopeptide[vii] derived bifunctional iminophosphophorane (BIMP)[viii] catalyst proved uniquely efficient at catalysing the enantioselective desymmetrisation. Equally key to the success of the desymmetrisation was the selection of the leaving group: too electron poor and the catalyst could not turn over, too electron rich and low conversion was observed. To our delight, ortho-nitrophenols provided the ideal balance of reactivity, enantioselectivity and phenolate basicity which would allow for the BIMP to re-enter the catalytic cycle. From a practical standpoint, the nitrophenol derived phosphonate esters were obtained by a one-pot sequence starting from commercially available or easily prepared alkyl phosphonate esters, phosphonic acids or phosphoryl dichlorides. Additionally, they proved to be exceptionally stable to both air and moisture, could be easily purified by silica gel chromatography and could be stored on the benchtop for months without any precautions.
Differently substituted ortho-phenols were found to be competent nucleophiles in the desymmetrisation, including those derived from the natural products thymol and totarol. Similarly, we found that variations of the R group on phosphorous were readily accommodated, with (hetero)aryl, alkyl and β-heteroatom substituents leading to the formation of the corresponding products with high yield and enantioselectivity (Figure 4). Importantly, the desymmetrisation step could be carried out on gram scale, providing ample enantioenriched intermediate for the second phase of the strategy, derivatisation by a subsequent, enantiospecific nucleophilic substitution.
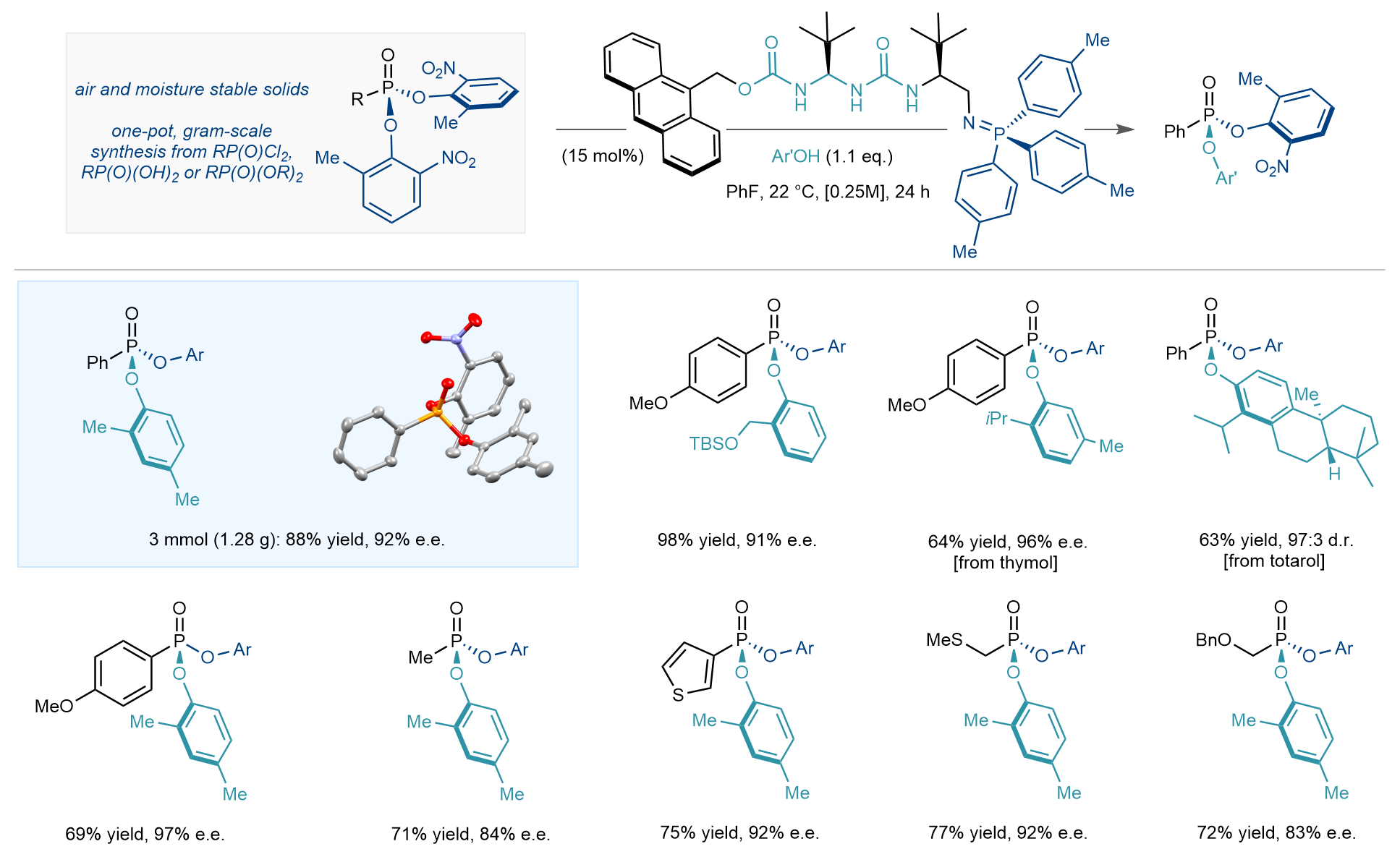
Figure 4. Select scope for the desymmetrisation stage: starting from air and moisture stable nitrophenol derived phosphonate esters (obtained by a one-pot synthesis from a variety of commercially available or readily prepared P(V) precursors) a range of enantioenriched P(V) products bearing (hetero)aryl and alkyl substituents were synthesized, enabled by BIMP catalysis. The products still possess a nitrophenol leaving group, poised for a second enantiospecific nucleophilic substitution.
Critically, the second nitrophenol leaving group could be easily displaced by a wide range of biologically relevant primary and secondary alcohol-based nucleophiles such as those derived from DNA and RNA nucleosides, also enabling the synthesis of an analogue of the antiviral drug sofosbuvir.[ix] Thiols and amines could also be used to displace the remaining nitrophenol enantiospecifically. In a related transformation, a substrate bearing a benzylic alcohol could be employed to effect the displacement intramolecularly providing access to an enantioenriched P(V) compound bearing the cycloSal prodrug motif.[x]
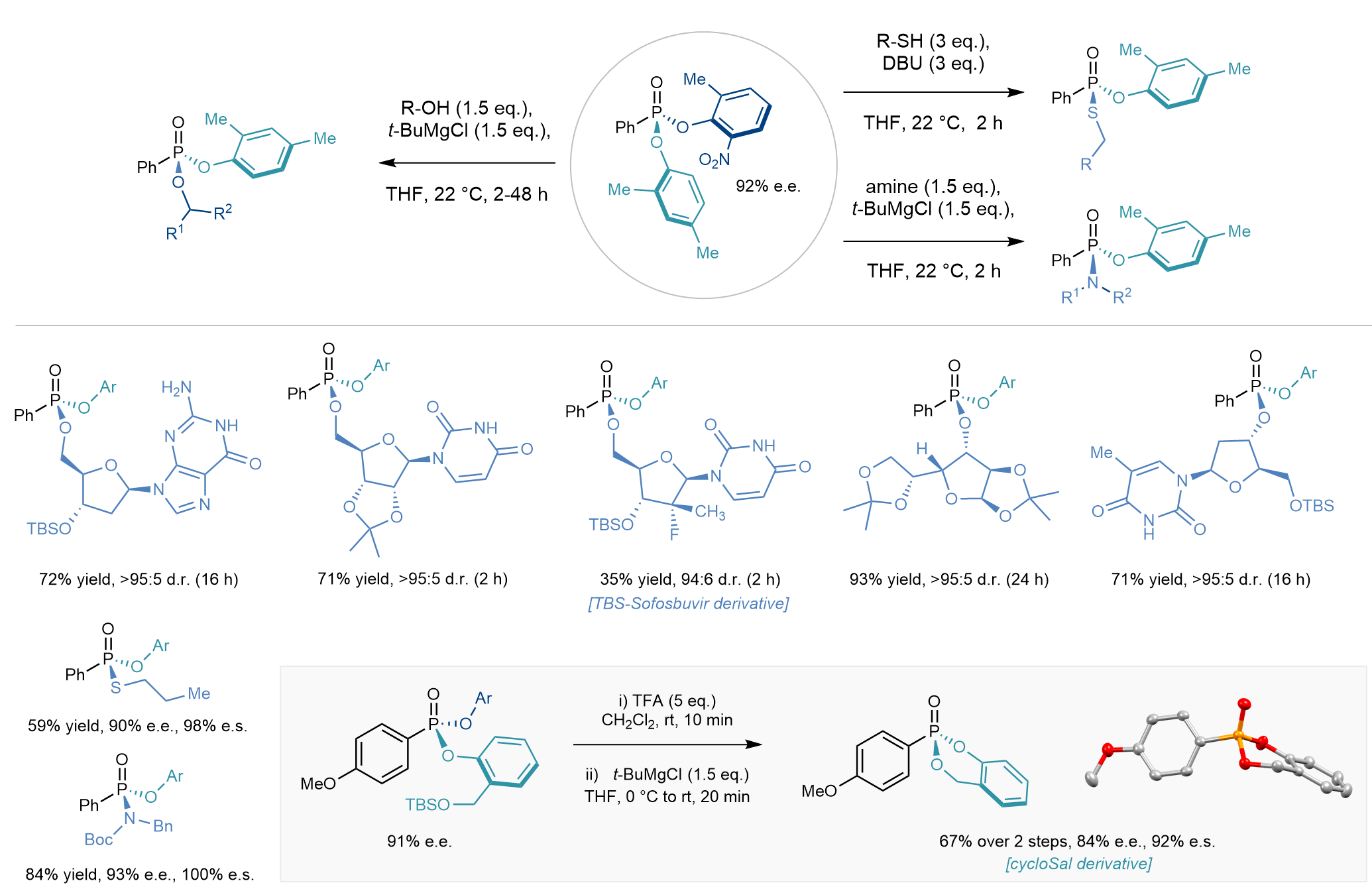
Figure 5. Select scope for the derivatization stage: from a single enantioenriched intermediate, diverse high value P(V) compound can be obtained with high enantiospecificity. Alternatively, nucleophilic displacement can occur intramolecularly affording compounds bearing the medicinally relevant cycloSal prodrug motif.
To gain insight into the origin of enantioselectivity for the desymmetrisation step, DFT studies were carried out, led by Dr. Ken Yamazaki of Okayama University. The computational studies revealed that addition of the phenol nucleophile to the P(V) electrophile is the rate- and enantio-determining step of the reaction with the formation of the pentacoordinate intermediate and elimination of the nitrophenol being energetically downhill. A detailed analysis of the non-covalent interactions between the catalyst and substrate in the transition state leading to the major enantiomer revealed the key role of the ureidopeptide H-bond donor in interacting with the nitro group on one of the leaving groups by an additional H-bond interaction which was absent in all other (less enantioselective) BIMP catalysts assessed which possessed more conventional urea and thiourea derived scaffolds.
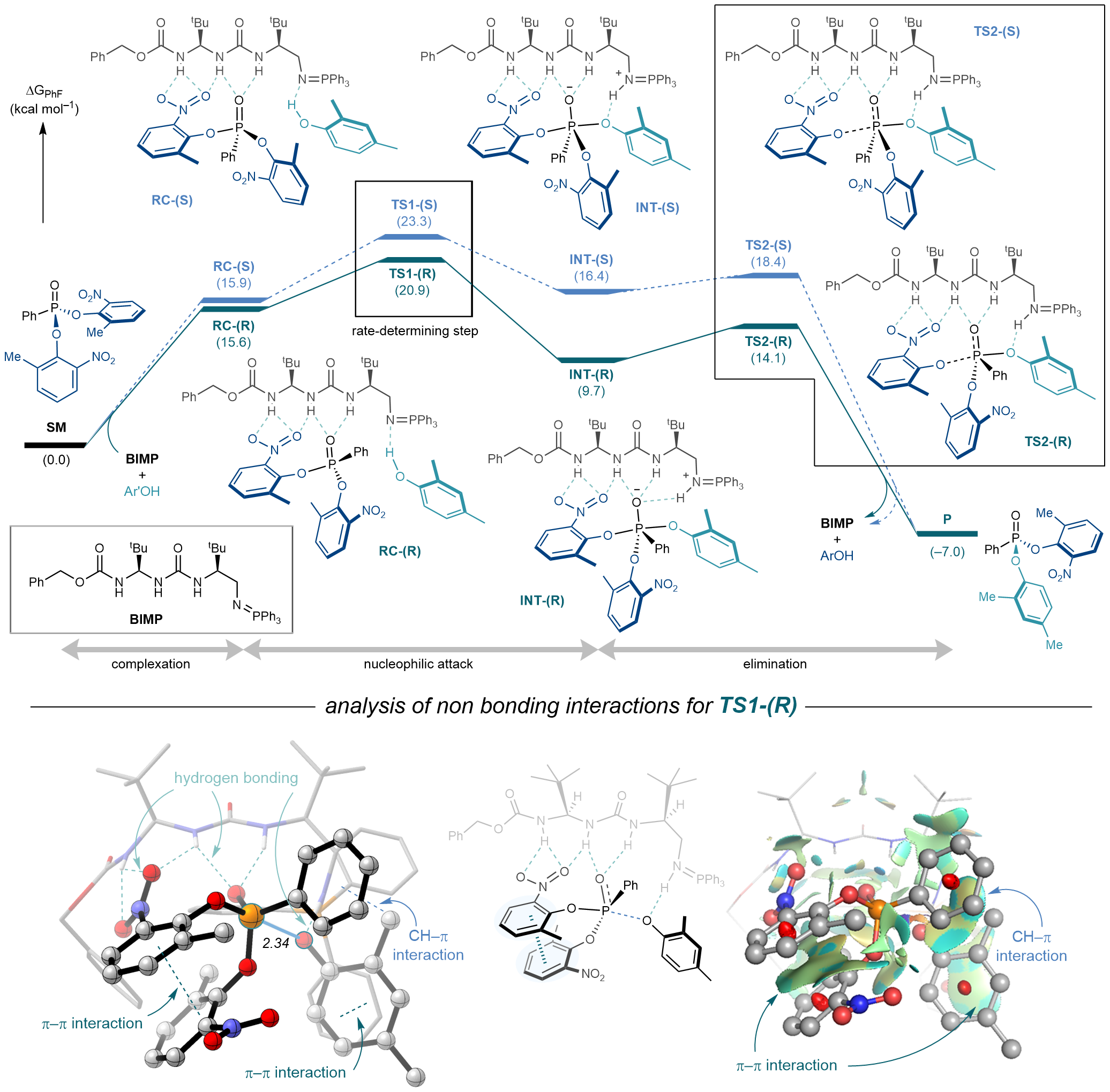
Figure 6. Computational studies revealed a plausible reaction pathway and elucidated the key non-bonding interactions between the ureidopeptide derived BIMP catalyst and the model substrate responsible for the high levels of enantioselectivity observed in the desymmetrisation by nucleophilic substitution.
Now possessing an arsenal of ureidopeptide BIMP catalysts at our disposal and guided by extensive computational studies we view this work as an open door to more general, efficient, and modular protocols for the synthesis of these valuable P(V) containing moieties. Efforts to render the enantioselective desymmetrisation amenable to a wider range of nucleophiles whilst generating even more versatile enantioenriched intermediates are well underway in our laboratories, we will be delighted to share our exciting findings in the coming months.
References
[i] Rodriguez, J. B. & Gallo-Rodriguez, C. The Role of the Phosphorus Atom in Drug Design. ChemMedChem 14, 190 –216 (2018).
[ii] Warren, T. K. et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016).
[iii] Wang, Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395, 1569–1578 (2020).
[iv] OMS. World health organization model list of essential medicines. Ment. Holist. Heal. Some Int. Perspect. 119–134 (2019).
[v] Lemouzy, S., Giordano, L., Hérault, D. & Buono, G. Introducing Chirality at Phosphorus Atoms: An Update on the Recent Synthetic Strategies for the Preparation of Optically Pure P-Stereogenic Molecules. European J. Org. Chem. 2020, 3351–3366 (2020).
[vi] Following the disclosure of this work as a preprint (Formica, M. et. al. Catalytic Enantioselective Nucleophilic Desymmetrisation of Phosphonate Esters. ChemRxiv, 2022 doi: 10.26434/chemrxiv-2021-5714s-v2), and while this manuscript was under revision an elegant and complementary catalytic enantioselective nucleophilic desymmetrisation of phosphoryl dichlorides was disclosed by the Jacobsen group employing anion binding catalysis: Forbes, K. C., Jacobsen, E. N. Enantioselective hydrogen-bond-donor catalysis to access diverse stereogenic-at-P(V) compounds. Science, 376, 1230 (2022).
[vii] Diosdado, S. et al. Catalytic Enantioselective Synthesis of Tertiary Thiols From 5 H -Thiazol-4-ones and Nitroolefins: Bifunctional Ureidopeptide-Based Brønsted Base Catalysis. Angew. Chemie Int. Ed. 52, 11846–11851 (2013).
[viii] Formica, M., Rozsar, D., Su, G., Farley, A. J. M. & Dixon, D. J. Bifunctional Iminophosphorane Superbase Catalysis: Applications in Organic Synthesis. Acc. Chem. Res. 53, 2235–2247 (2020).
[ix] Simmons, B., Liu, Z., Klapars, A., Bellomo, A. & Silverman, S. M. Mechanism-Based Solution to the ProTide Synthesis Problem: Selective Access to Sofosbuvir, Acelarin, and INX-08189. Org. Lett. 19, 2218–2221 (2017).
[x] Meier, C. cycloSal phosphates as chemical trojan horses for intracellular nucleotide and glycosylmonophosphate delivery - Chemistry meets biology. Eur. J. Org. Chem. 1081–1102 (2006).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in