A chromosome-wide view of an exceptionally diverse helminth
Published in Ecology & Evolution

Helminths, more commonly called parasitic worms, represent an incredibly diverse group of organisms that exploit a variety of life-history strategies for their survival and persistence. Parasitic worms often cause significant sickness and can cause death to their hosts. More than a billion people worldwide are infected with at least one species of parasitic worm, and in response to this, they are the target of some of the world’s largest drug delivery campaigns to control them as a public health burden. They are also a major problem for our domesticated animals and represent a significant animal health and economic challenge, particularly on our livestock industries.
A major concern regarding the control of parasitic worms is the evolution of anthelmintic drug resistance. While the emergence of resistance by human-infective helminths is less well established, resistance is clearly established in helminths of veterinary importance throughout the world, such that in some regions, treatment options are running out. Understanding the mechanisms by which these worms become resistant is a hot research topic; deciphering this mystery may provide new ways to diagnose and monitor resistance emergence so that parasites can be more effectively managed and the lifespan of the limited number of drugs currently available can be preserved, as well as developing new ways to control them. The gastrointestinal worm, Haemonchus contortus, is one such parasite of livestock ruminants that is globally distributed and has rapidly evolved resistance to all drug classes used to control it. Commonly called the Barber’s pole worm, due to its distinctive blood-filled gut of the female parasite that spirals around the paler reproductive tract throughout most of its body (Figure 1), it feeds on its host’s blood causing symptoms associated with anaemia and at high parasite burdens, infections can be fatal. H. contortus is also a “model parasite” commonly used in drug and vaccine development research (one of the few parasite species for which a commercially available vaccine is in use), attractive due to the fact that it can be cultured, preserved, and can be experimentally mated (i.e., different strains can be genetically crossed within the host) and manipulated (e.g., RNA interference, in vitro drug assays).
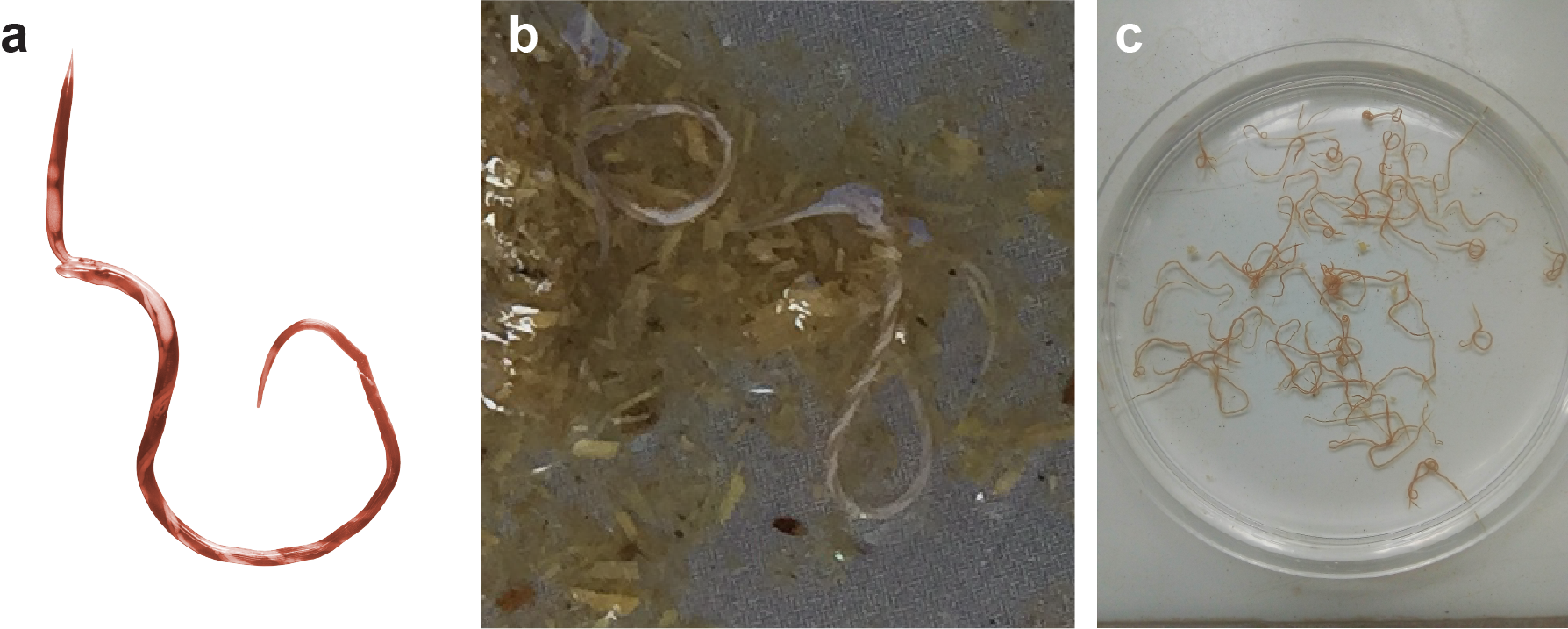
Figure 1. a. A single adult female H. contortus, showing its distinctive “barber’s pole” gut (courtesy of John Gilleard, University of Calgary, Canada). b. Two H. contortus worms, freshly isolated within gut contents (courtesy of Stuart Ralph, University of Melbourne, Australia). c. Many H. contortus worms, cleaned and ready to be used in the lab (courtesy of Essia Sebai, University of Tunis El Manar, Tunisia).
We have recently completed the chromosome-scale genome assembly of H. contortus, now published in Communications Biology. A major milestone of the BUG Consortium (https://bugconsortium.wordpress.com/), a multidisciplinary group focused on building genomic resources to map genes driving anthelmintic resistance, these genome resources represent years of work by many people, including long-running collaborative efforts of the Parasite Genomics Group at the Wellcome Sanger Institute (https://www.sanger.ac.uk/group/berriman-group/), the University of Glasgow, and the Moredun Research Institute. Motivated by the utility of the draft genome and transcriptome published in 2013 for investigating H. contortus biology (see Laing et al, https://doi.org/10.1186/gb-2013-14-8-r88), we quickly realised that a highly-contiguous genome assembly was necessary to understand precisely how drug selection shapes genetic variation throughout the genome. However, at the time, we did not appreciate the effort that was required to resolve the genetic complexities of such a small but genetically diverse invertebrate. Taking a “let’s try everything” approach (which appreciably is not a common approach afforded by many outside of a major genomics institute), we applied a barrage of short- and long-read high throughput sequencing technologies; the major challenge to this approach was that large amounts of DNA was required for this sequencing, which meant pooling hundreds to thousands of these genetically diverse worms, making assembling the genome extremely complicated. By integrating these multiple approaches together with extensive manual curation, we were able to “thread a path” through this variation to achieve chromosomal-scale assembly scaffolds (Figure 2a). This new chromosomal perspective has provided us with insight into chromosome evolution between closely related species (Figure 2b), improved representation of gene annotations and transcriptional diversity, and a more precise understanding of genome- and population-wide genetic diversity (Figure 2c). The genome assembly and its associated annotation resources have been made available at the main repository for helminth genomics data, WormBase Parasite (https://parasite.wormbase.org/Haemonchus_contortus_prjeb506/Info/Index/), and will be continued to be improved over time for the benefit of the research community.
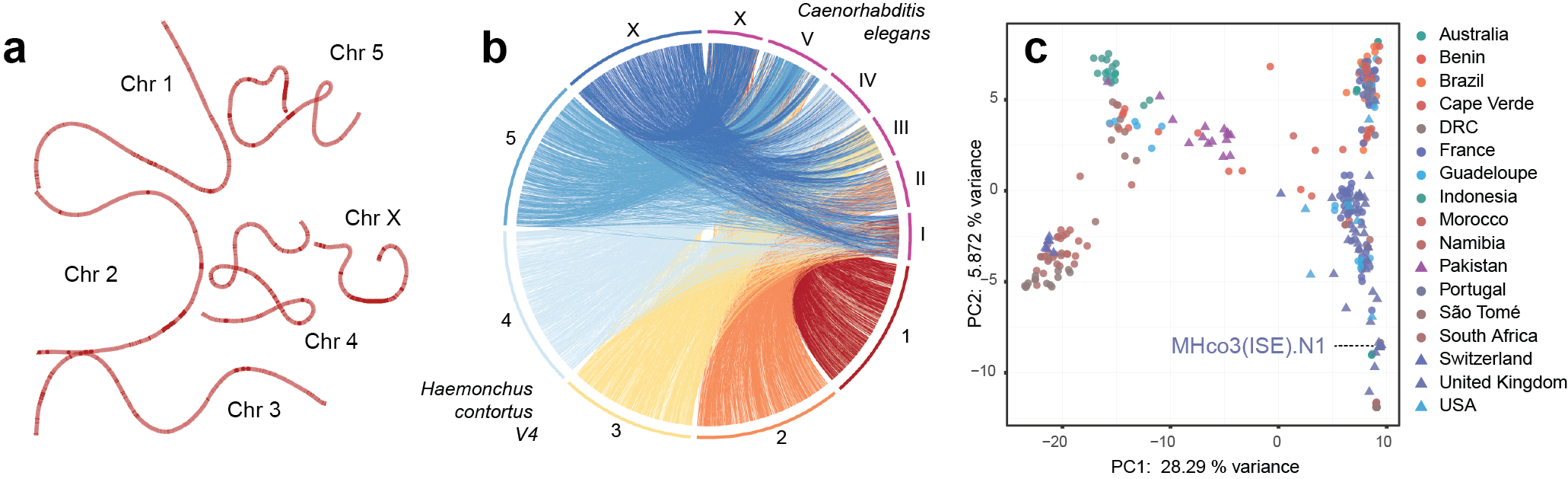
Figure 2. a. Genome-graph representation of the five autosomal and one sex-linked scaffold of H. contortus. b. CIRCOS plot showing shared sequences between the chromosomes of H. contortus and the model free-living nematode, Caenorhabditis elegans. More than 80% of the genes are conserved between chromosomes, but their order is almost completely rearranged. c. Global diversity of almost 350 H. contortus isolates, including genetic placement of the MHco3(ISE).N1 strain used for the genome assembly.
Mapping genetic variation associated with drug resistance still remains a major challenge and an active research area for us. However, the chromosomal perspective of this genome resource has greatly improved our understanding of drug-mediated selection on the genome, allowing us to map discrete regions that contain variants likely driving drug resistance. It will be exciting to see how these resources will be used in the future to better understand the complex but intriguing lives of parasitic worms.
If you have any further questions about the work or use of these resources, feel free to send me an email (stephen.doyle[at]sanger.ac.uk) or reach out on Twitter (@stephenrdoyle).
Follow the Topic
-
Communications Biology

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.
Related Collections
With Collections, you can get published faster and increase your visibility.
From RNA Detection to Molecular Mechanisms
Publishing Model: Open Access
Deadline: May 05, 2026
Signalling Pathways of Innate Immunity
Publishing Model: Hybrid
Deadline: May 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in