Why is asymmetric synthesis important? The three-dimensional structure of molecules can play an important role in their biological, chemical and physical properties. Molecules which have the same atom-to-atom connectivity but have their atoms arranged differently in space are referred to as stereoisomers. Stereoisomers can be further classified as either enantiomers or diastereomers. Enantiomers are stereoisomers which are mirror images of one another but are non-superimposable. Diastereomers, in contrast, are not mirror images of one another. Asymmetric synthesis refers to the synthesis of a specific enantiomer of a given target compound. As enantiomers often behave differently in biological systems, it is generally necessary to prepare enantiomerically pure drug substances.
Background to the discovery: In 2012, Lawrence and co-workers accomplished the total synthesis of the related Incarvillea natural products (±)-incarviditone and (±)-incarvilleatone through a biomimetic dimerization of (±)-rengyolone. The rengyolone used in this process is racemic —a 1:1 mixture of enantiomers — represented in Fig. 1 as blue and red structures for clarity. Through analysis of the relative configuration of incarviditone, it is clear that it arises from a homochiral dimerization of rengyolone, where the red and blue enantiomers only react with themselves and not each other. As enantiomers will behave identically in the absence of additional chiral information in the system, the probability of red-red dimerization is equal to that of blue-blue dimerization, resulting in racemic incarviditone. An asymmetric synthesis of incarviditone could therefore be achieved if an enantiopure sample of rengyolone were used in place of the racemate.
The picture becomes significantly more complicated when trying to design an asymmetric synthesis of the other natural product, incarvilleatone, which is a heterochiral dimer (i.e., blue-red and red-blue). Again, there is an equal likelihood that the red or blue enantiomer will act as the nucleophile in the initial oxa-Michael reaction, with the opposite enantiomer acting as the electrophile, resulting in a 1:1 mixture of both enantiomers of the final product. In order to preferentially make a specific enantiomer of incarvilleatone, one would have to bias the reaction in favour of either the red or blue enantiomer of rengylone acting as the nucleophile and the opposite enantiomer acting as the electrophile. This type of process, where a racemic sample of a precursor molecule is transformed into a single enantiomer of a product, is known as an enantioconvergent reaction.
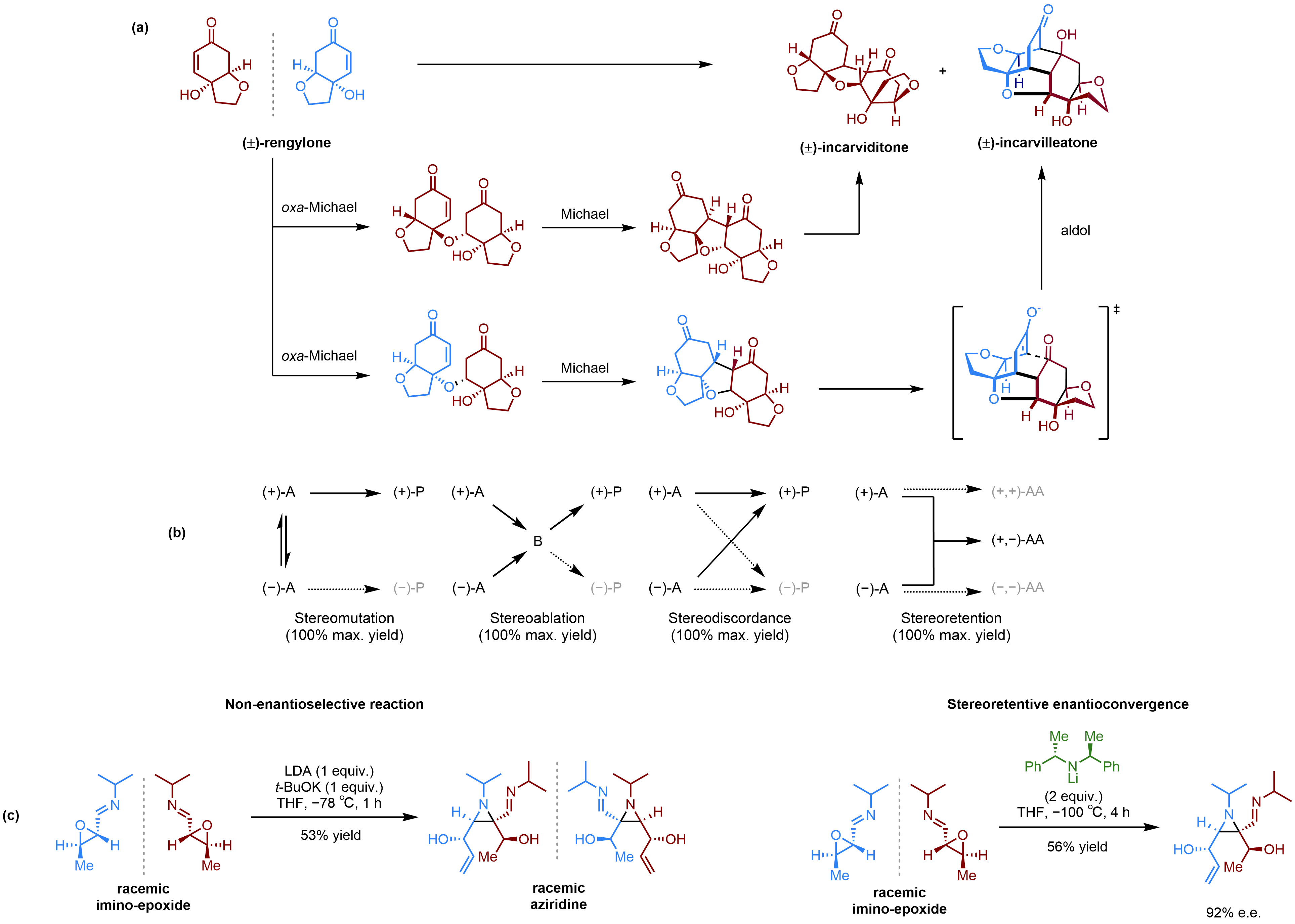
Figure 1: (a) Dimerization of (±)-rengyolone to form (±)-incarviditone and (±)-incarvilleatone. Only one enantiomer of (±)-incarviditone and (±)-incarvilleatone is shown for clarity. (b) Enantioconvergent reaction templates, highlighting the key difference between stereoretention and the previously established templates. (c) The first stereoretentive enantioconvergent reaction – an aza-Darzens reaction mediated by a chiral lithium amide base.
Enantioconvergent reactions: Prior to our report in Nature Chemistry, there were three established ways of achieving enantioconvergence — stereomutation, stereoablation and stereodiscordance (a term which we introduce for the first time in our report). These three approaches differ based on the fate of each of the starting enantiomers, but crucially all involve the loss of some stereochemical information.
Revisiting the problem of how to make incarvilleatone through an enantioconvergent approach, we recognised that it would not neatly fall into any of these previously established strategies, as we would need to retain the configuration of both starting enantiomers. This new type of stereoretentive enantioconvergence would therefore represent a conceptually new way of making chiral molecules in enantiopure form.
Inspired by this train of thought, our group scoured the literature for examples of highly selective heterochiral dimerization reactions with a view to establishing this new strategy as experimentally viable. We were particularly taken by an unusual aza-Darzens reaction reported by Würthwein and co-workers because of the stereochemical complexity of the aziridine product. Through judicious choice of chiral base, we were able to successfully modify Würthwein’s approach and realise our first example of a stereoretentive enantioconvergent reaction, accessing the aziridine product in 92% e.e..
Multicomponent approach: Reports of heterochiral selective dimerization reactions are quite rare in the literature. We, therefore, became curious to see if we could apply our concept in a different type of reaction where we could more easily control the heterochiral selectivity. To this end, we designed an approach centred on the use of bifunctional symmetry-breaking linkers, which would connect the two enantiomers of the racemate together to form a non-meso product. Conceptually, one end of the linker would react selectively with the faster reacting enantiomer through a kinetic resolution (KR). Subsequently, the unreacted functional group of the linker, a spectator until then, would undergo a second coupling with the remaining starting material enantiomer to give the heterochiral dimer. Not only does this approach allow us to easily dial-in heterochiral selectivity but it also benefits from a statistical amplification of enantiopurity, meaning that even with moderate selectivity in the initial KR products will be obtained in a high level of enantiopurity.
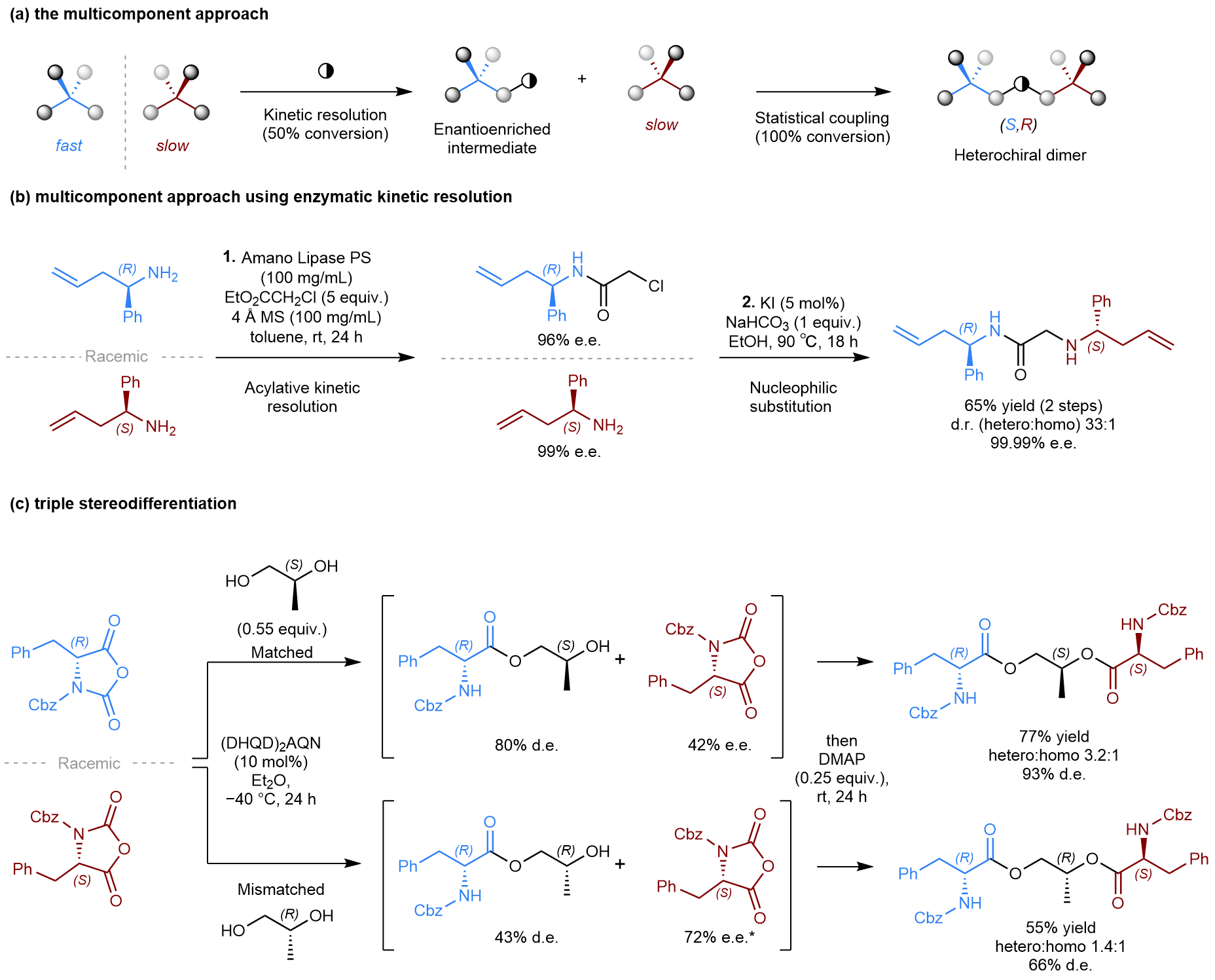
Figure 2: The multicomponent approach. (a) Design of the multicomponent approach. (b) The multicomponent approach using enzymatic kinetic resolution. (c) Exploiting triple stereodifferentiation and exploring matched/mismatched effects.
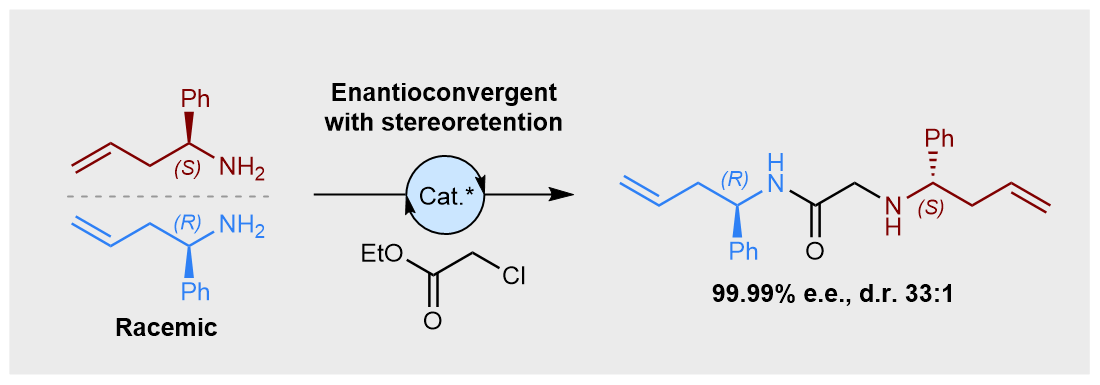
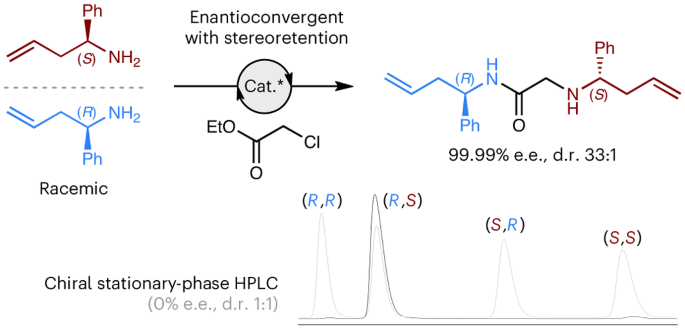
This multicomponent strategy was attempted on several known KR reactions, using a variety of established tactics in asymmetric synthesis (i.e., chiral auxiliary, biocatalysis, and organocatalysis). We were pleased to see that the concept was transferable across this set of reactions, using a bis‑electrophilic linker design, delivering the heterochiral dimers in high levels of enantiopurity in each case (>97% e.e.). The promising potential of this approach is clearly shown in the biocatalytic acylation of homoallylic amines using ethyl chloroacetate as the linker. Thanks to the statistical enantioamplification, and a highly selective initial kinetic resolution, the final hetereochiral product is formed in virtually enantiopure form (99.99% e.e.). To our delight, the unwanted homochiral dimer was only observed in trace amounts, only observable by HPLC (hetero:homo = 33:1).
Finally, we decided to push the concept a little further, attempting a diastereoconvergent multicomponent transformation. In this case, an enantiopure bis-nucleophilic linker was used, which forced us to consider substrate-driven diastereocontrol. For this attempt, a racemic amino acid derivative (a Leuchs' anhydride) was chosen with propylene glycol as a chiral unsymmetrical diol. Evaluation of the two opposite enantiomers of the diol led to the identification of a matched linker that afforded the final heterochiral product in 93% d.e., while the mismatched analogue resulted in a significantly lower 66% d.e.. This demonstrates the importance of the absolute configuration of the linker on the selectivity. This is an example of triple stereodifferentiation, as the selectivity depends on the absolute configuration of all three components; the reacting enantiomer, the linker and the organocatalyst.
We envisage this new approach to asymmetric synthesis will be particularly interesting to organic chemists as it will allow for new types of chiral molecule to be prepared in enantiopure form for the first time. Some potential applications could be in the drug design space, where accessing all possible stereoisomers of new types of chiral molecule is of paramount importance. There are also a number of natural products which arise from heterochiral dimerization reactions, such as incarvilleatone (previously mentioned) or kingianin D, which could potentially be prepared in enantiopure form through stereoretentive enantioconvergence. Furthermore, one could envisage utilising this type of approach to access a variety of supramolecular structures.
At its core, this work underpins a fundamentally new way of thinking about how we stitch molecules together to access enantiomerically pure products. The paucity of intrinsically heterochiral-selective dimerization reactions raises interesting questions about the origin of such selectivity. We actually believe this can be partly rationalised because of self-imposed hesitance, as researchers would previously avoid dimerizing racemic samples due to the complex issues of homochiral-heterochiral selectivity that we are interested in. Nevertheless, in our experience, heterochiral dimerization processes are challenging to predict, and therefore design. We hope that our work enables further such curiosity-driven exploration of this phenomenon among the synthetic community. It is exciting to think where this curiosity may lead us. For example, there is an emerging appreciation for the role that heterochiral selectivity may have played in the origin of biological homochirality — underpinned by the recent work of Blackmond and co-workers.1
1 Deng, M., Yu, J. & Blackmond, D.G. Symmetry breaking and chiral amplification in prebiotic ligation reactions. Nature 626, 1019–1024 (2024). https://doi.org/10.1038/s41586-024-07059-y
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in