Adiabatic versus Non-Adiabatic Electron Transfer at 2D Electrode Materials
Published in Chemistry


Electrochemical systems have at least three common features: electron transfer (ET) from a donor to an acceptor, an electrochemical interface, and ability to change kinetics and thermodynamics by applying an electrode potential. Combining and modifying these basic constituents forms the basis for a wide variety of applications in electrocatalysis, energy storage and electrosynthesis, and also for deeper understanding of the deceptively simple-looking ET processes that underlie many branches of chemistry and physics. In fact, all three aspects are highly complex at the atomic scale, precluding precise control and insight. ET is a quantum mechanical process and can involve complications from long-range tunnelling or non-adiabaticity. It proceeds either with or without strong interactions between the electron acceptor and donor. The electrified interface is a very complex reaction environment, where the structure and properties differ greatly from those of the constituent bulk phases, and are strongly influenced by the electrode potential, which also affects the ET mechanism, thermodynamics, and kinetics.
Disentangling this complexity requires the combination of precise experiments on well-defined electrochemical interfaces with advanced atomic simulations and pen-and-paper theory. This is the approach taken in our contribution, where we have developed an integrated experiment-simulation-theory approach shown in Figure 1 of the article (reproduced below) to address one of the most basic electrochemical questions: is an outer-sphere electron transfer reaction (OS-ET) with weak interactions between the electrode and electron acceptor adiabatic or non-adiabatic? Previous microscopic understanding of OS-ET was largely based on idealized theoretical models that typically did not capture the complexity of electrochemical interfaces, simulations that did not account for the electrode potential, and electrochemical measurements that provided limited datasets and insufficient kinetic resolution. The consequence has been a hitherto unsurmountable gap between theory and experiment.
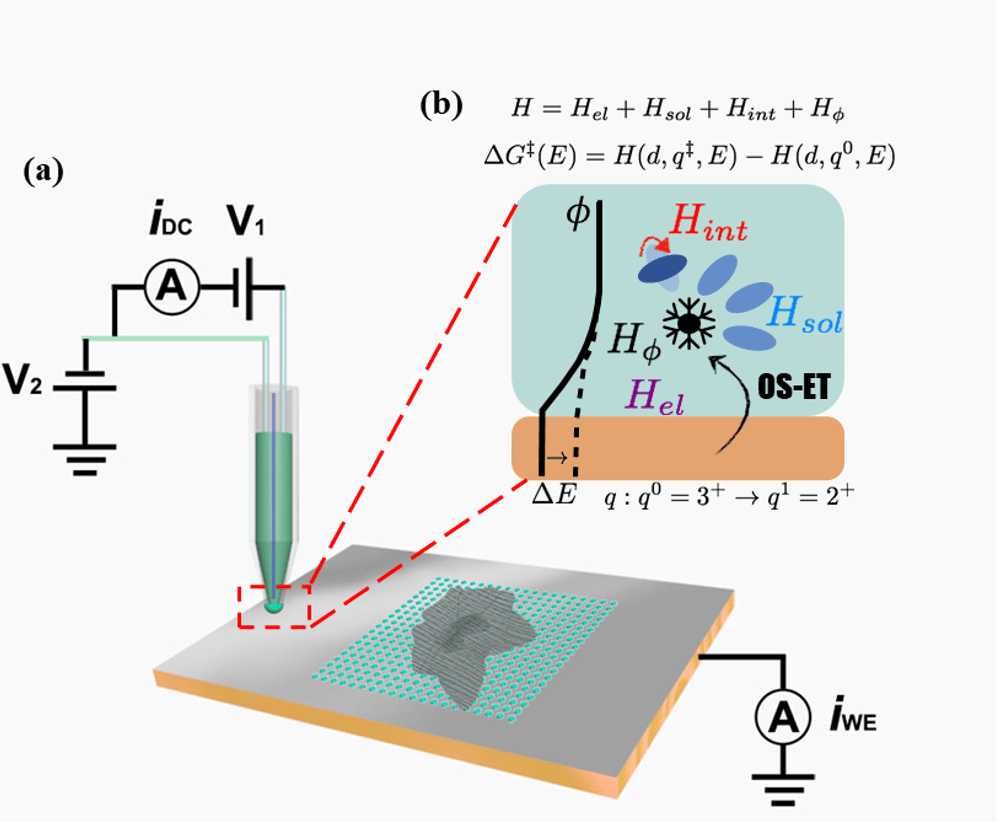
Figure 1. (a) shows a schematic SECCM setup used in our work and (b) shows the microscopic interactions of OS-ET used in the model Hamiltonian.
To close this gap, we measured the ET kinetics of a classical OS-ET redox couple, [Ru(NH3)6]3+/2+, on well-defined graphene-functionalized copper electrodes with a varying number of graphene layers. This electrode configuration is of considerable interest for studying electrochemistry and electrocatalysis at a wide range of metal-supported 2D materials. By using scanning electrochemical cell microscopy (SECCM), we were able to measure the OS-ET kinetics as a function of the number of graphene layers and show that the OS-ET rate decreased as the number of graphene layers is increased. The OS-ET rate was found to be about four times larger on the monolayer than on the bilayer graphene-Cu electrode. This observation was difficult to understand using previous knowledge; adiabatic OS-ET kinetics are usually considered to be independent of the electrode material whereas a non-adiabatic process would have a much stronger dependency on the electrode.
Usually a computational/theoretical chemist working on electrochemical problems needs to make difficult compromises to reach a suitable, simplified model of the complex system studied experimentally. The graphene/copper electrode provided an ideal platform for a detailed and integrated computational and theoretical study: getting a chance to work on an experimentally well-defined system with nanoscale resolution is a rare treat. We modelled the graphene-modified copper electrodes with grand canonical density functional theory (GC-DFT) to explicitly account for the effect of the electrode potential on properties of the electrochemical interface. These computational results were then used for parametrizing an extended model Hamiltonian describing OS-ET kinetics in terms of chemically or physically transparent parameters.
The GC-DFT + model Hamiltonian -approach proved to be a very fruitful approach in explaining the experimental observations. We were able to convincingly demonstrate that OS-ET can be rationalized within an adiabatic framework, where graphene layers act as ‘distance buffer’, producing an increase in the effective barrier to ET due to an increased electrostatic potential resulting from the addition of more graphene layers. This was an unexpected finding as previous experimental and theoretical works have largely neglected the effect of the electrostatic potential on OS-ET.
Our work provides tangible evidence for the complementary insight and benefits from the integration of experiments, simulations, and theory to understand electrochemical reactions. This allowed us to clarify the microscopic mechanism of an electrochemical reactions and its dependency on the three main features of an electrochemical system – ET, interface, and the electrode potential – from experimental, computational, and theoretical perspectives. The study also shows the value of studying model systems and advancing nanoscale experiments, constant potential DFT, and even basic ET theory to obtain microscopic understanding and control on fundamental electrochemical processes.
Read more in the Nature Communications paper: https://www.nature.com/articles/s41467-021-27339-9
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in