Beyond the Black Box: Explainable Chemical Artificial Intelligence From Real Space Quantum Chemistry
Published in Chemistry, Physics, and Computational Sciences

Background
Traditionally, the rigorous study of chemistry has been limited to small molecules due to the high computational costs involved. This changed dramatically with the advent of Artificial Intelligence (AI) models capable of predicting a wide range of chemical descriptors and properties without the need for running expensive quantum mechanical calculations. Recently, AI models have rapidly advanced, enabling the computational simulation of complex and large molecules closer to the systems tackled by state-of-the-art research in Chemistry.
However, most chemical AI models face two major challenges. Firstly, they often display limited extrapolation abilities, which can result in erratic predictions when applied to previously unexplored domains of the chemical space. Secondly, they are uninterpretable, providing accurate estimates without explaining the underlying logic, thus behaving as black boxes.
Quantum Chemical Topology
To address this problem, we propose leveraging the rigor of modern physical theories, particularly Quantum Chemical Topology (QCT). This theory is founded on the topological analysis of various scalar fields, such as electron density, enabling the decomposition of real space into a collection of basins with well-defined local properties. As such, QCT provides an ideal foundation for developing advanced theoretical tools for chemistry, including the Quantum Theory of Atoms in Molecules (QTAIM) and the Interacting Quantum Atoms (IQA) energy partitioning scheme.

Thus, we believe that by creating AI architectures designed to learn local QCT properties, we can develop interpretable and transferable models. This strategy contrasts with the common approach in the field, which often involves directly predicting molecular properties as a sum of independent terms that frequently lack physical significance.
Transferability and Interpretability
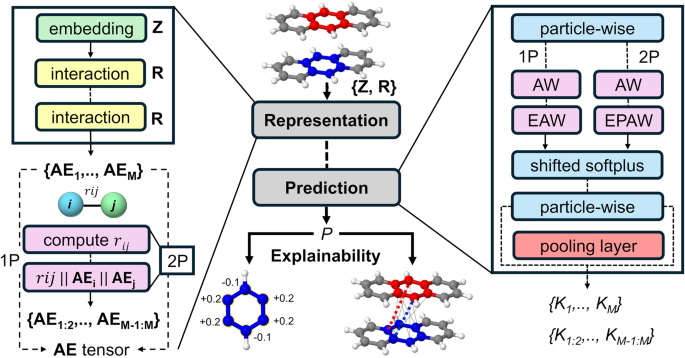
The architecture developed, called SchNet4AIM, relies on SchNet to autonomously describe the chemical environments of each atom embedded in a molecule, which are then used to establish a correspondence between structure and property spaces. In this way, molecular quantities are reconstructed as a combination of atomic and pairwise contributions. Such an approach contrasts with what is commonly done in the field, which often employs terms lacking well-defined physical meanings to obtain the final predictions.
Our results reveal that this approach yields highly accurate and physically consistent atomic and pairwise predictions. However, the true innovation lies not solely in accuracy but in the broad applicability and adaptability of the models. SchNet4AIM demonstrates proficiency in predicting highly complex molecules beyond its training dataset, a departure from the typical limitations seen in many chemical AI models, known as extrapolation challenges. This unique capability, stemming from the transferability and robustness of QCT theories, contributes to addressing a major hurdle in contemporary chemical AI: the quest for universally applicable models that transcend accuracy constraints.
Furthermore, the utilization of chemical locality leads to an unforeseen outcome: the models exhibit inherent interpretability. Our findings indicate that SchNet4AIM can discern and elucidate the electronic delocalization accompanying supramolecular binding processes. Consequently, it not only identifies the emergence of chemical contacts but also highlights the predominant pairwise interactions driving such binding. This marks a crucial advancement in the pursuit of chemically explainable AIs (XCAIs), an emergent area within AI research.
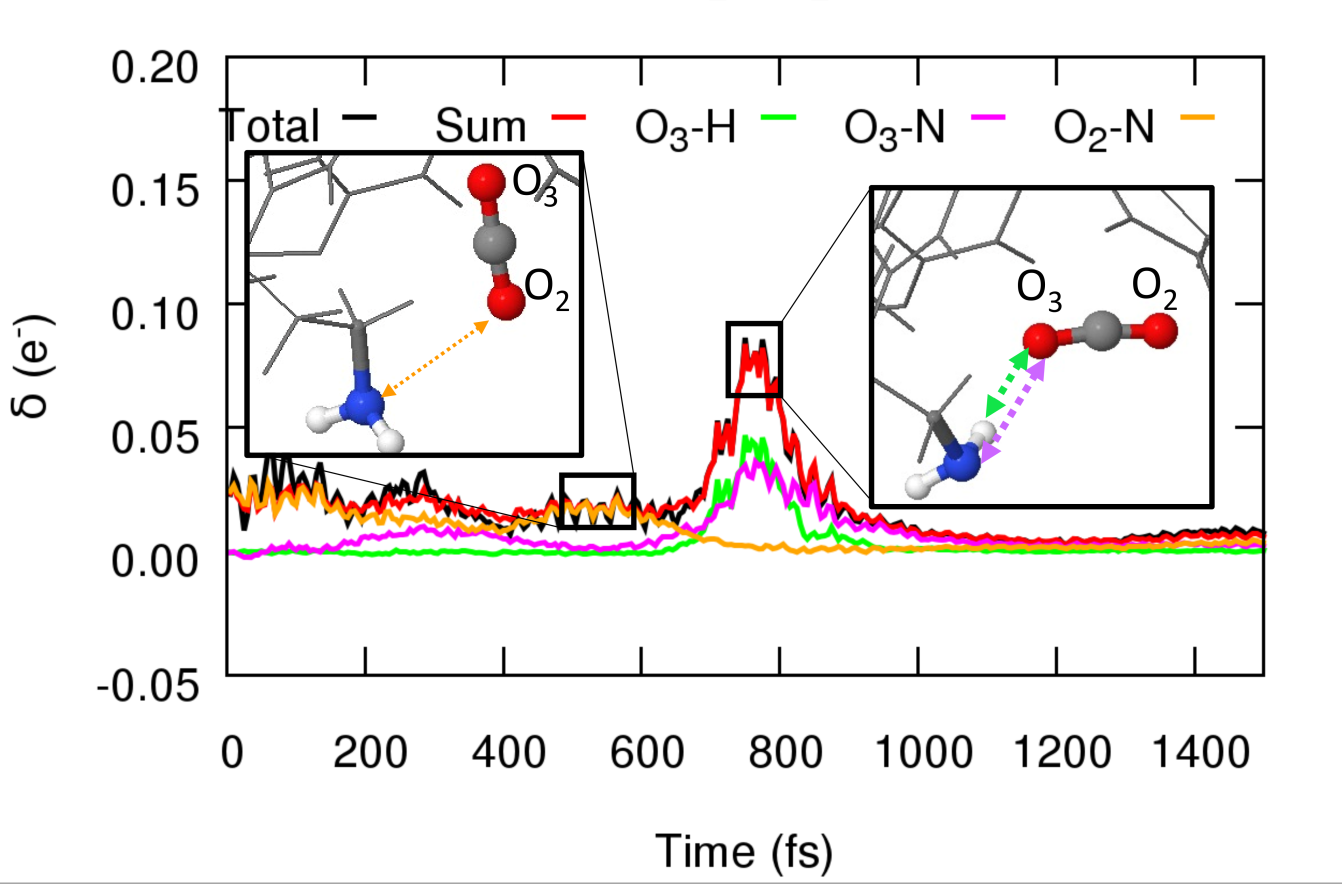
Evolution of the electronic delocalization, as forecasted by SchNet4AIM, during the formation of a supramolecular complex between a ligand and a receptor. Each distinct color represents the (explained) prevailing pairwise components elucidated by SchNet4AIM.
Future Work
This breakthrough heralds an exciting convergence between Quantum Chemical Topology and Artificial Intelligence, paving the way for the development of computational tools with vast potential applications. That said, SchNet4AIM encounters certain challenges, including comprehending how molecular properties are actually affected by the physical variables of the system.
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Ask the Editor – Space Physics, Quantum Physics, Atomic, Molecular and Chemical Physics
Got a question for the editor about Space Physics, Quantum Physics, Atomic, Molecular and Chemical Physics? Ask it here!
Continue reading announcementRelated Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in