Beyond the fusion gene: uncovering an RNA surveillance checkpoint in leukemia
Published in Biomedical Research
 When I first entered the field of hematological malignancies, I did not arrive as a classical leukemia biologist. My scientific training and instinct were rooted in RNA biology and RNA therapeutics. I was used to thinking about transcripts as dynamic molecular entities: how they are spliced, exported, translated, surveilled, degraded, and eventually converted into biological outcomes. This way of thinking shaped how I looked at disease genes: not only as DNA lesions or protein-coding units, but as RNA molecules with their own life histories.
When I first entered the field of hematological malignancies, I did not arrive as a classical leukemia biologist. My scientific training and instinct were rooted in RNA biology and RNA therapeutics. I was used to thinking about transcripts as dynamic molecular entities: how they are spliced, exported, translated, surveilled, degraded, and eventually converted into biological outcomes. This way of thinking shaped how I looked at disease genes: not only as DNA lesions or protein-coding units, but as RNA molecules with their own life histories.
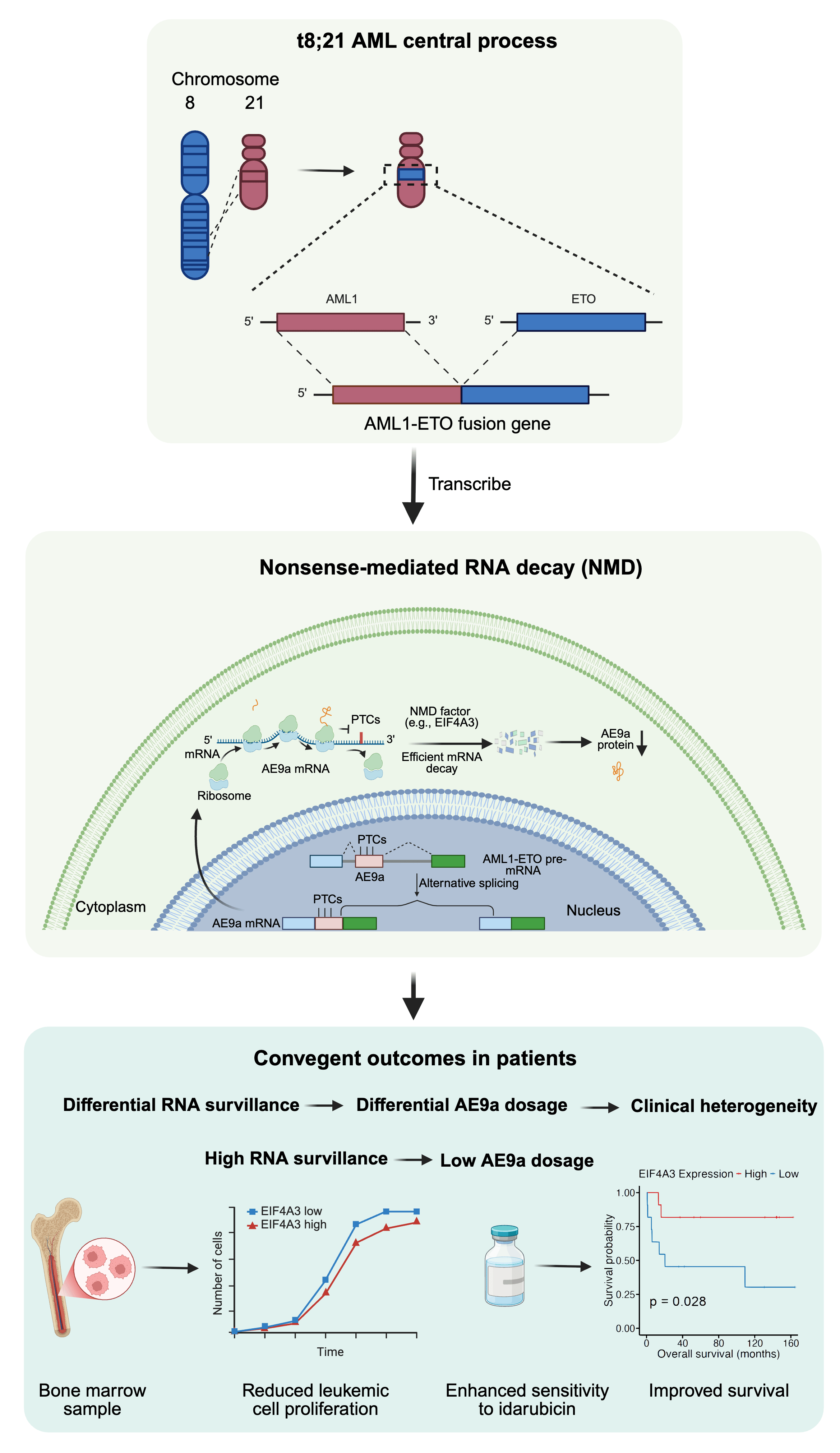
That perspective became especially important when I began to study t(8;21) acute myeloid leukemia (AML), a subtype of AML defined by a chromosomal translocation between chromosomes 8 and 21. In this translocation, the RUNX1 (AML1) gene on chromosome 21 becomes fused to the RUNX1T1 (ETO) gene on chromosome 8, creating the AML1-ETO fusion gene, also known as RUNX1-RUNX1T1. The fusion joins the DNA-binding Runt homology domain of AML1 to nearly full-length ETO, changing the behavior of AML1 as a transcription factor and disrupting normal hematopoietic differentiation. For many years, much of the field has understandably focused on how AML1-ETO rewires downstream signaling, transcriptional repression, chromatin state, epigenetic regulation, self-renewal, and myeloid maturation. These are central questions in leukemia biology.
But as an RNA biologist, I found myself asking a slightly different question: before an oncogenic fusion product changes downstream pathways, how is that fusion product itself generated, processed, surveilled, and controlled?
This question became even more intriguing when I learned about AML1-ETO9a, or AE9a. AE9a is an alternatively spliced, truncated isoform of AML1-ETO generated by inclusion of an additional ETO9a cassette exon. Previous studies had shown that AE9a has strong leukemogenic activity and can accelerate leukemia development in experimental models. Yet in human t(8;21) AML, AE9a expression varies markedly among patients. Why do some patients accumulate more AE9a than others? Is AE9a abundance simply a byproduct of alternative splicing noise, or is there an active cellular mechanism that buffers its level?
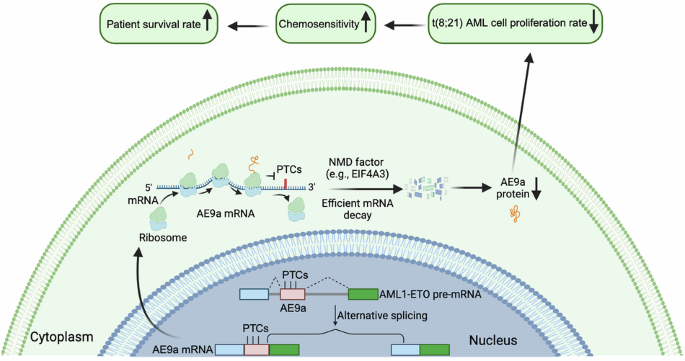
The clue came from looking carefully at the RNA architecture of the AE9a isoform. When the ETO9a cassette exon is included, the resulting transcript contains premature termination codons, or PTCs. To many people, this might look like a technical detail in the transcript map. To an RNA biologist, it immediately raises a red flag: this transcript could be recognized by nonsense-mediated mRNA decay (NMD). NMD is a conserved RNA surveillance pathway that detects and degrades transcripts carrying premature stop codons. It is often introduced in textbooks as a quality-control system for eliminating aberrant RNAs. But in disease biology, NMD can do much more. It can reshape transcript dosage, influence cell fitness, alter immune recognition, and modify therapeutic response.
This observation led to the central hypothesis of our study: perhaps AE9a is not merely produced by alternative splicing, but is continuously restrained by NMD. If the NMD machinery is strong in a given patient’s leukemic cells, AE9a may be efficiently degraded. If NMD capacity is weaker, AE9a may escape surveillance, accumulate, and promote leukemic growth.
To test this idea, we first analyzed RNA-seq data from primary t(8;21) AML CD34⁺ hematopoietic stem and progenitor cells. We could detect ETO9a exon inclusion in patient samples, confirming that the PTC-containing AE9a transcript is indeed generated in primary leukemic cells. More importantly, AE9a inclusion was inversely correlated with the expression of multiple NMD-related factors, including UPF1, UPF3B, SMG1, SMG7, DHX34, and EIF4A3. Among these, EIF4A3 and DHX34 showed particularly strong negative correlations with AE9a inclusion. This was an exciting moment, because it suggested that patient-to-patient variation in AE9a abundance might reflect differences in RNA surveillance capacity rather than simply differences in total fusion-gene expression.
The next challenge was to move from correlation to mechanism. We used pharmacological approaches to perturb translation-coupled NMD and direct NMD components. In t(8;21) AML cell lines such as Kasumi-1 and SKNO-1, inhibition of NMD increased ETO9a-containing transcripts. Nuclear-cytosolic fractionation further showed that this increase occurred mainly in the cytoplasmic fraction, consistent with the idea that AE9a transcripts are made in the nucleus, exported to the cytoplasm, and then selectively eliminated by translation-coupled NMD. At the protein level, disrupting SMG1 or EIF4A3 increased AE9a accumulation, indicating that NMD controls not only AE9a RNA abundance but also the effective dosage of the oncogenic AE9a protein.
A particularly important part of the work was validating this mechanism in primary patient cells. This was also where collaboration became essential. I am especially grateful to my co-corresponding author and clinical collaborator, Prof. Li Yu, whose expertise in hematological malignancies helped anchor this RNA-biology question in the clinical reality of t(8;21) AML. His support and perspective were important for connecting our mechanistic hypothesis to patient-derived cells, disease heterogeneity and therapeutic response. In CD34⁺ hematopoietic stem and progenitor cells and CD34⁻ leukemic fractions from t(8;21) AML patients, pharmacological inhibition of NMD increased ETO9a/AE9a levels. Genetic depletion of NMD factors, including EIF4A3 and DHX34, produced convergent effects. These experiments were technically demanding because primary AML patient cells are precious, variable, and not always easy to manipulate. But they were essential. Without patient-cell validation, the story would have remained a cell-line mechanism. With patient-cell validation, we could begin to see NMD as a physiological buffer of AE9a dosage in human t(8;21) AML. I am also grateful to all co-authors and collaborators who helped move this project from an RNA-biological observation to a patient-relevant leukemia study.
The most surprising result came when we looked at patient survival. EIF4A3 is often discussed in cancer as a pro-tumorigenic factor, and in broader AML contexts it has been proposed as a potential therapeutic target. However, biology is context-dependent. In our t(8;21) AML cohort, high EIF4A3 expression was associated with significantly improved overall survival. This association was not observed in non-t(8;21) AML or in other major cytogenetic AML groups. This specificity was critical. It suggested that EIF4A3 is not simply “good” or “bad” in AML; rather, its effect depends on which transcripts are under its surveillance. In the AE9a-producing context of t(8;21) AML, EIF4A3-dependent NMD appears to act as a protective dosage-control mechanism.
We then asked whether increasing EIF4A3 could functionally restrain leukemia-related phenotypes. EIF4A3 overexpression reduced AE9a RNA and protein in Kasumi-1 and SKNO-1 cells. It also slowed t(8;21) AML cell proliferation, while not comparably suppressing healthy CD34⁺ progenitor expansion under our experimental conditions. Importantly, EIF4A3 overexpression enhanced sensitivity to idarubicin, a chemotherapy agent used in AML treatment. These findings connected RNA surveillance to three clinically relevant layers: oncogenic fusion-isoform dosage, leukemic proliferation, and chemotherapy response.
Looking back, the most rewarding part of this project was not only identifying a new mechanism, but reframing how we think about fusion-driven leukemia. A fusion gene is not the end of the story. Between a chromosomal translocation and a leukemic phenotype lies a long molecular journey: transcription, splicing, RNA export, surveillance, translation, protein stability, and functional embedding in cellular state. Our study highlights one point along that journey—NMD-mediated buffering of AE9a—as a disease-modifying checkpoint.
For me personally, this project also reinforced the value of entering a field from a different angle. Coming from RNA biology and RNA therapeutics, I may not have initially viewed leukemia in exactly the same way as researchers trained primarily in hematologic malignancy signaling, transcription, or clinical cytogenetics. But that difference became productive. It allowed us to ask why an oncogenic fusion isoform accumulates in some patients and not others, and whether RNA surveillance could help explain clinical heterogeneity.
The broader implication is that post-transcriptional regulation may represent an underappreciated layer of fusion-oncogene biology. In t(8;21) AML, AE9a is a potent leukemogenic isoform, but its dosage is not fixed. It is actively shaped by the cell’s RNA quality-control machinery. Preserving or restoring such surveillance mechanisms may one day complement existing approaches that target downstream signaling, chromatin dependencies, or chemotherapy response.
This paper began with a simple RNA-biological observation: an oncogenic fusion isoform contains premature termination codons. Following that observation led us from transcript architecture to patient heterogeneity, from RNA surveillance to leukemic fitness, and from EIF4A3 to chemotherapy sensitivity. For us, that journey captures the spirit of “Behind the Paper”: sometimes the most important clue is hidden not in what a gene does downstream, but in what happens to its RNA before it ever becomes a protein.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in