Building a robust tool for processing metagenomes, one error at a time
Published in Microbiology
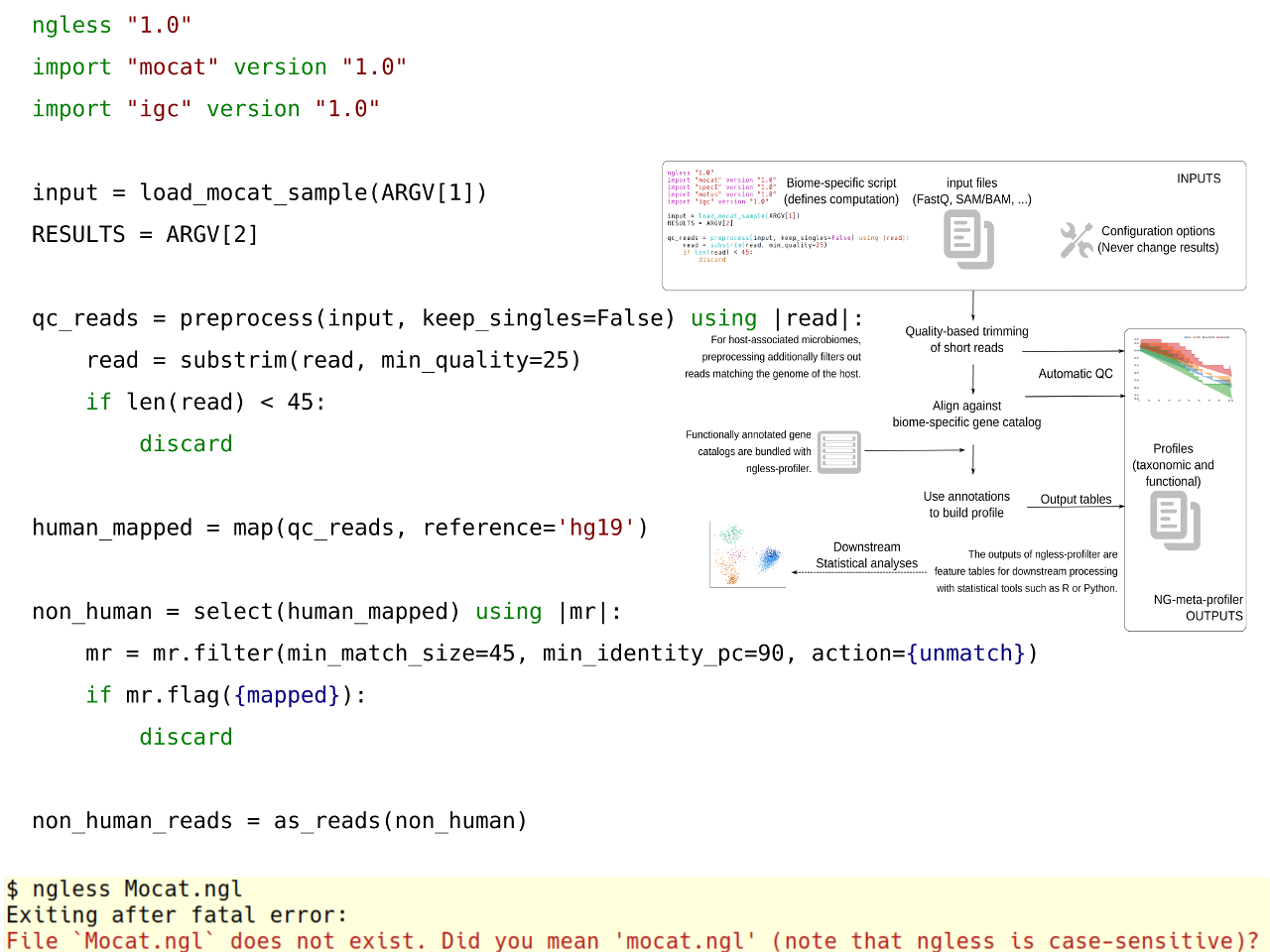

The goal of NG-meta-profiler is to take metagenomes as inputs and produces estimates of either taxonomic or functional abundances. At a high-level, we use a well-validated approach to metagenomic analysis: use a pre-annotated gene catalogue as a reference against which to align metagenomic reads [1-3]. The big innovation is in the way the tool was designed and is presented to the users.
What do we want from our computational tools?
On the one hand, we want standard pipelines that embody best practices and enable comparisons between different studies and datasets, we want them to be easy to install and use by individuals without a strong background in bioinformatics. On the other hand, we want the flexibility to adapt to the specificity of each dataset and expert bioinformaticians will want to tweak and combine tools to address different contexts and requirements.
We propose a solution to these seemingly conflicting goals by presenting a set of standard pipelines that can be easily adapted while maintaining reproducibility. The central concept behind the work is a novel programming language for processing NGS data called NGLess which was used to write these standard pipelines.

NG-meta-profiler and NGLess are just now being published as a journal article, but the tools themselves have been in development for several years and were always available as open-source software. In fact, the initial intuition that this could be a good idea first came to me back in 2012! Looking back at my mail archives, I found an email from me to co-author Ana Teresa Freitas from September 21st 2012 with some initial thoughts on the idea as a follow-up to a conversation we had earlier.
Then, the project proceeded through three phases:
Proof-of-concept (2013-2014).
Internal use, maturation (2015-2016).
Release to wider public (2017-present).
The initial goal was to design a simple programming language and write a piece of software that would solve some toy problems. This work was the bulk of Paulo Monteiro’s MS Thesis, which he submitted at the end of 2014.
At this point, most of the ideas were in place and we had proved that the concept could work to process data, but the tool was slow and would often produce hard-to-understand error messages. After improving the performance, we started using the tool internally for our own projects (e.g., [4]). The NGLess interpreter became more robust with time and started to produce better error messages. This was a slow process as behind each little improvement there was often a confusing debugging session. For example, a malformed FastQ file could cause an “Out of memory” error. Naturally, we would then attempt to run the analysis in a large-memory machine before realizing what was going on.

Around 2017, we started encouraging others to use the tools and receiving feedback. We realized that the way the software was built caused issues with some types of HPC clusters (even though it had run fine on our systems). We received further reports of confusing error messages and improved those. We realized that there were some inconsistencies that confused new users and streamlined the interface.
Individually, all of these were minor changes, but collectively these hundreds of small improvements make all the difference between the finicky proof-of-concept we had in 2014 and a tool we could proudly recommend to others. So, in 2018, we finally decided it was time to submit it as a manuscript to a journal, and you can now read about it at Microbiome.
REFERENCES
[1] Kultima JR, Coelho LP, Forslund K, Huerta-Cepas J, Li SS, Driessen M, Voigt AY, Zeller G, Sunagawa S, Bork P. MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics. 2016 Apr 8;32(16):2520-3.
[2] Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular biology and evolution. 2017 Aug 1;34(8):2115-22.
[3] Ugarte A, Vicedomini R, Bernardes J, Carbone A. A multi-source domain annotation pipeline for quantitative metagenomic and metatranscriptomic functional profiling. Microbiome. 2018 Dec;6(1):149.
[4] Schmidt TS, Hayward MR, Coelho LP, Li SS, Costea PI, Voigt AY, Wirbel J, Maistrenko OM, Alves RJ, Bergsten E, de Beaufort C. Extensive transmission of microbes along the gastrointestinal tract. eLife. 2019 Feb 12;8:e42693.
Follow the Topic
-
Microbiome
This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.

Related Collections
With Collections, you can get published faster and increase your visibility.
Oncobiome
This collection of papers delves into the burgeoning field of oncobiome research, exploring the intricate relationship between cancer and the microbiome. The oncobiome encompasses the diverse microbial communities residing in and on the human body, which influence cancer development, progression, and treatment responses. By examining these interactions, our aim is to unravel the complex mechanisms through which the microbiome impacts oncogenesis and therapeutic outcomes.
This compilation highlights cutting-edge research, offering insights into potential diagnostic markers and novel therapeutic strategies, thereby advancing our understanding of cancer biology and paving the way for innovative, microbiome-targeted cancer treatments.
This is a cross-journal collection between:
Experimental Hematology and Oncology
Articles will undergo the standard peer-review process of the journal to which they are submitted and are subject to either the BMC editorial policies or those of BJC Reports. Articles will be added to the Collection as they are published. The Editors have no competing interests with the submissions which they handle through the peer review process. The peer review of any submissions for which the Editors have competing interests is handled by another Editorial Board Member who has no competing interests.
Publishing Model: Open Access
Deadline: Ongoing
Animal Gut Nutrition and Greenhouse Gas Mitigation
Animal Microbiome, Journal of Animal Science and Biotechnology and Microbiome call for submissions to the collection on Animal Gut Nutrition and Greenhouse Gas Mitigation.
Efforts to reduce greenhouse gas emissions from livestock systems increasingly hinge on innovations in animal gut nutrition. The dynamic relationship between the gut microbiome and nutrient utilization plays a pivotal role in shaping methane output, feed efficiency, and overall sustainability. Advances in microbial ecology—particularly in understanding the role of gut microbiome in nutrient metabolism—are opening new pathways for mitigating emissions while enhancing productivity. These developments support the implementation of climate-smart agricultural strategies to address climate change and its impacts.
Looking ahead, continued research in this field has the potential to yield innovative solutions such as targeted probiotic supplementation, which could further optimize gut function and enhance nutrient absorption. These advancements may lead to reduced greenhouse gas emissions while improving animal health and productivity. By deepening our understanding of the animal gut microbiome, we can contribute significantly to sustainable agricultural practices that benefit both the environment and food security.
We invite researchers to contribute to this special Collection on Animal Gut Nutrition and Greenhouse Gas Mitigation. Topics of interest include but are not limited to:
- Animal Gut Microbiome and Feed Efficiency
- Greenhouse Gas Mitigation Strategies
- Rumen Fermentation Dynamics
- Nutrient Utilization in Livestock
- Probiotic Supplementation Effects
- Sustainable Livestock Production Practices
- Climate-Smart Agriculture Innovations
This Collection supports and amplifies research related to SDG 13, Climate action.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Animal Microbiome fees, Journal of Animal Science and Biotechnology fees, Microbiome fees). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 04, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in