CapTrap-Seq: A platform-agnostic and quantitative approach for high-fidelity full-length RNA sequencing
Published in Protocols & Methods and Genetics & Genomics


Unlocking the Full Potential of Long-Read RNA Sequencing with CapTrap-seq
Long-read RNA sequencing has revolutionized the annotation of eukaryotic genomes by providing comprehensive insights into full-length RNA transcripts. Despite significant advancements, achieving complete and reliable transcript identification remains challenging. Addressing this gap, our team developed CapTrap-seq, a novel cDNA library preparation method that integrates the Cap-trapping strategy for selecting transcript beginnings with the oligo(dT) strategy for selecting their ends.
CapTrap-seq: A New Method for Comprehensive Transcript Identification
CapTrap-seq offers a dual-selection approach that enhances the accuracy and completeness of RNA transcript libraries1. By leveraging the Cap-trapping technique, it captures the 5' end of RNA molecules, while the oligo(dT) strategy secures the 3' ends. This method ensures the inclusion of full-length transcripts, crucial for accurate genome annotation (Figure panel 1. CapTrap-seq Workflow).
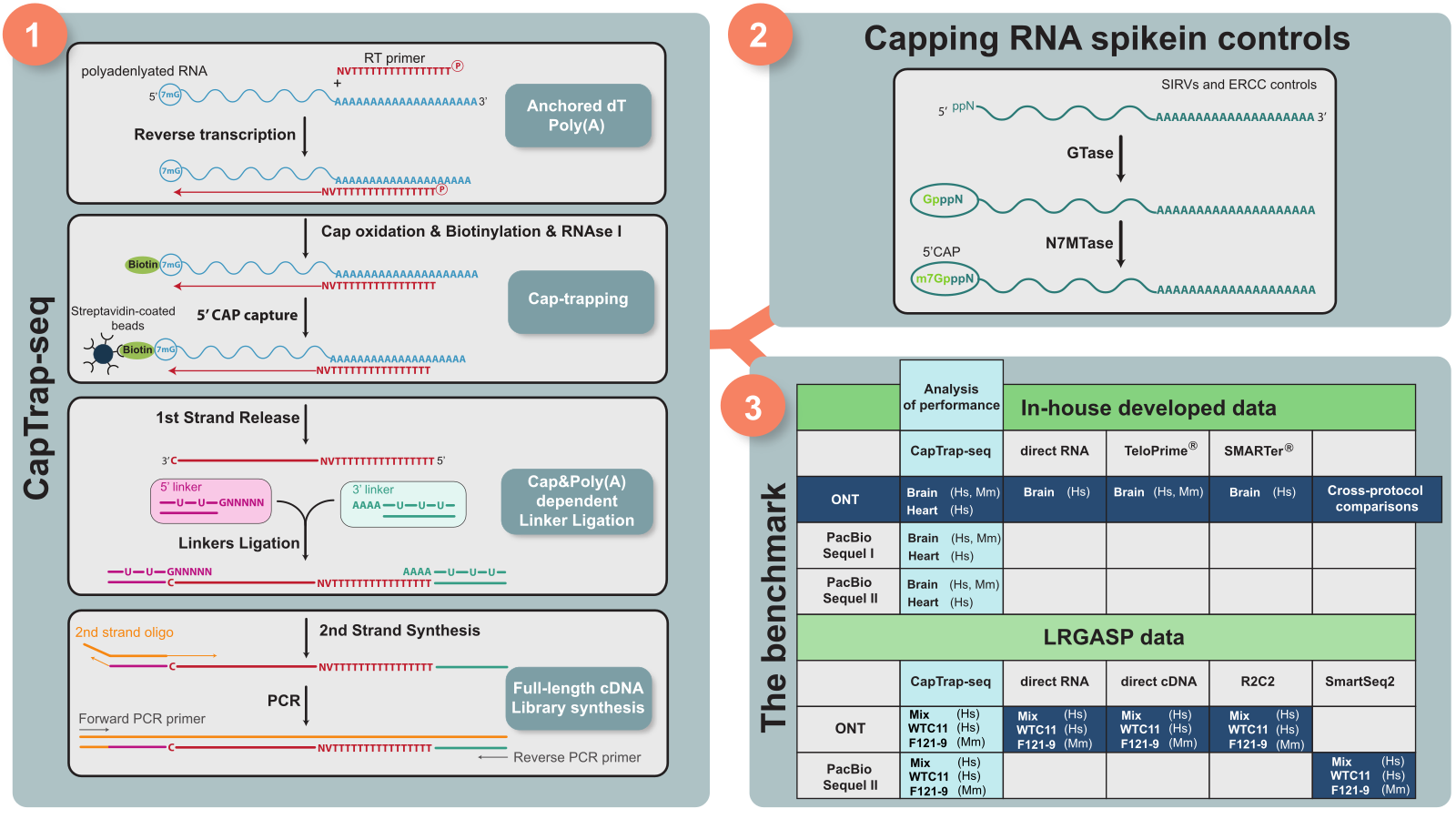
Evaluating the Performance of CapTrap-seq
Our study assessed the performance of CapTrap-seq using human and mouse tissues, by comparing it with other RNA-seq library preparation methods across two sequencing technologies: Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio). A novel capping strategy for synthetic RNA sequences was implemented mimicking natural cap structures to test the quantitative and completeness capabilities of CapTrap-seq (Figure panel 2. Capping RNA Spike-in Controls)2.
Results from the LRGASP Consortium
The Long-read RNA-seq Genome Annotation Assessment Project (LRGASP) Consortium rigorously evaluated CapTrap-seq3. The results highlighted its ability to produce full-length transcript sequences across different platforms. CapTrap-seq demonstrated competitive performance in transcript reconstruction and quantitative accuracy, producing highly reproducible data.
Platform Independence and High-Throughput Efficiency
CapTrap-seq stands out due to its platform-independent nature and high-throughput efficiency. It effectively filters out uncapped nucleic acids, reducing contamination from genomic DNA and ribosomal RNA. This makes it particularly suitable for generating high-confidence isoform-specific transcript models.
Benchmarking Against Other Protocols
Compared to other protocols, CapTrap-seq consistently produced a higher proportion of full-length transcripts, especially on the ONT platform (Figure panel 3. Benchmark Tables).
Applications and Future Directions
CapTrap-seq is already contributing to the GENCODE project, where thousands of its transcript models are being integrated into the gene set. Additionally, we developed an enzymatic capping strategy for synthetic RNA spike-in controls, enhancing CapTrap-seq sensitivity, quantitativeness, and accuracy.
Challenges and Limitations
Despite its advantages, CapTrap-seq has some limitations. It requires a substantial amount of starting RNA (5μg) and involves a complex laboratory procedure, which may lead to shorter RNA molecules compared to standard methods. While no specific length bias was observed for ERCC and SIRV controls, CapTrap-seq tends to produce shorter reads. Additionally, the protocol primarily targets polyadenylated RNA molecules, necessitating modifications to accommodate non-polyadenylated fractions.
Conclusion
CapTrap-seq represents a significant advancement in long-read RNA sequencing, offering a robust and versatile method for generating accurate full-length transcript maps. As genome sequencing projects expand, CapTrap-seq could play a pivotal role in providing the high-quality transcription data essential for precise genome annotations.
Acknowledgements
I gratefully acknowledge the invaluable assistance and support provided by all the authors of this manuscript. Special thanks are extended to Barbara Uszczynska-Ratajczak, Roderic Guigó, and their respective labs for their collective discussions and guidance, which have been instrumental in refining the methodology described in the Nature Communications manuscript. I am deeply grateful for all their invaluable contributions to this project.
Blog images credit to Barbara Uszczynska-Ratajczak.
Bibliography
- Carbonell-Sala, Sílvia & Guigó Roderic. CapTrap-Seq cDNA library preparation for full-length RNA sequencing, 24 May 2024, PROTOCOL (Version 1) available at Protocol Exchange [https://doi.org/10.21203/rs.3.pex-2646/v1]
- Carbonell-Sala, Sílvia & Guigó, Roderic. 5' Capping protocol to add 5’ cap structures to exogenous synthetic RNA references (spike-ins), 24 May 2024, PROTOCOL (Version 1) available at Protocol Exchange [https://doi.org/10.21203/rs.3.pex-2649/v1]
- Pardo-Palacios, F. J. et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat. Methods (2024) doi:10.1038/s41592-024-02298-3.
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in