DiffModeler: large macromolecular structure modeling for cryo-EM maps using a diffusion model
Published in Chemistry, Protocols & Methods, and Cell & Molecular Biology
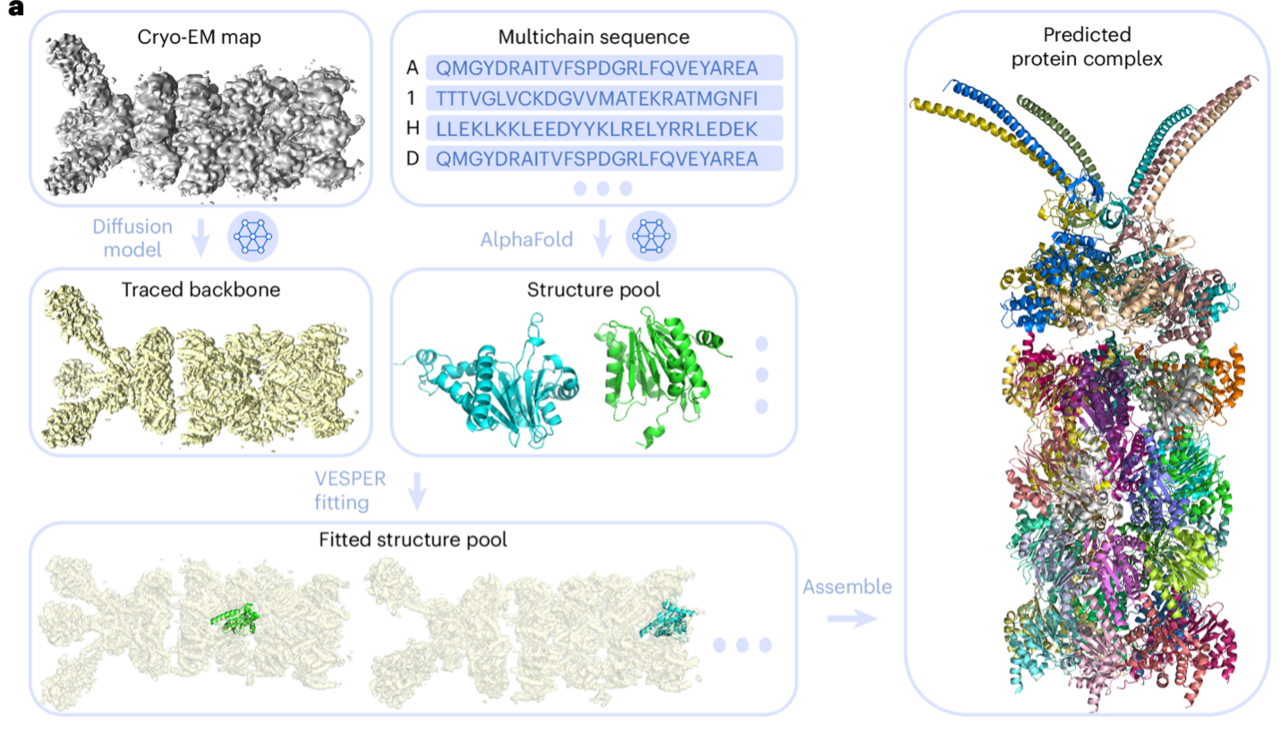
Proteins are essential biomolecules that perform a myriad of functions in living organisms, including enzymatic catalysis, cellular signaling, and molecular transport. Among experimental techniques for determining protein structures, cryogenic electron microscopy (cryo-EM) is now widely used as it can determine complex macromolecular structures that are challenging by conventional methods. Despite its advancements, modeling protein complex structures from cryo-EM maps remains a significant challenge, primarily due to limitations in map resolution and the inherent complexity of protein complexes. Particularly, direct tracing of main chain of proteins is very challenging when map resolution is worse than 5 Å. To this end, DiffModeler was developed for automated accurate protein complex structure modeling from cryo-EM maps with a primary focus on a middle to low resolution range, 5 Å to 10 Å and even worse.
The Kihara Lab is an interdisciplinary research group affiliated with both in biology and computer science (CS) departments. Our lab physically locates in the structural biology building, and we have observed numerous successful structural biology projects through cryo-EM in the last decade. We also worked on the cryo-EM related software development for more than six years. We have developed several tools for structure modeling (CryoREAD, MAINMAST, MAINMAST-Seg, DeepMAINMAST), structure detection (Emap2sec, Emap2sec+), structure evaluation (DAQ), structure refinement (DAQ-Refine) and map alignment (VESPER). They are all freely available on our EM-Server.
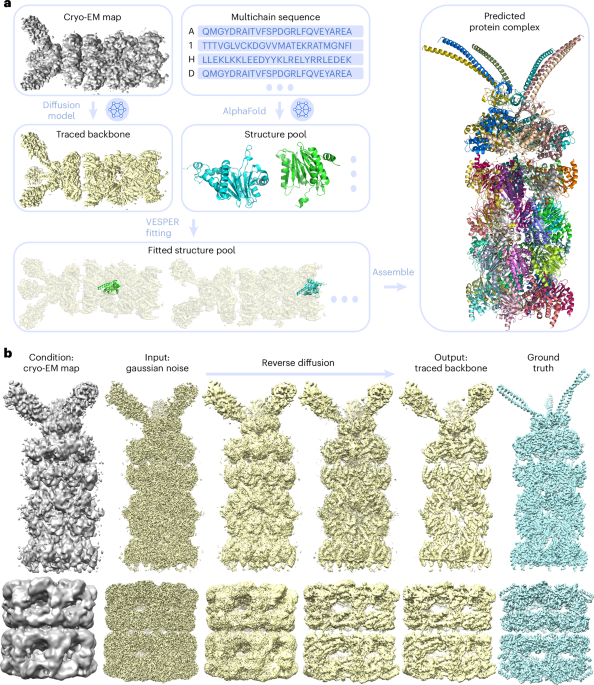
The new method, DiffModeler, uses diffusion model to automatically build a full protein complex structure by taking known structures or predicted models such as by Alphafold2 (AF2) as input. Specifically, it comprises four major steps: First, it detects the protein backbone positions in the input cryo-EM map by enhancing the map using a trained diffusion model. Second, it conducts the modeling of single-chain protein structures using AF2, where we can also use native structure if available. Third, single-chain structure models are fitted to the enhanced map using another tool developed in our lab, VESPER. Last, it selects and combines fitted single-chain poses to build the complete protein complex structures within the map. The overall framework is presented below.
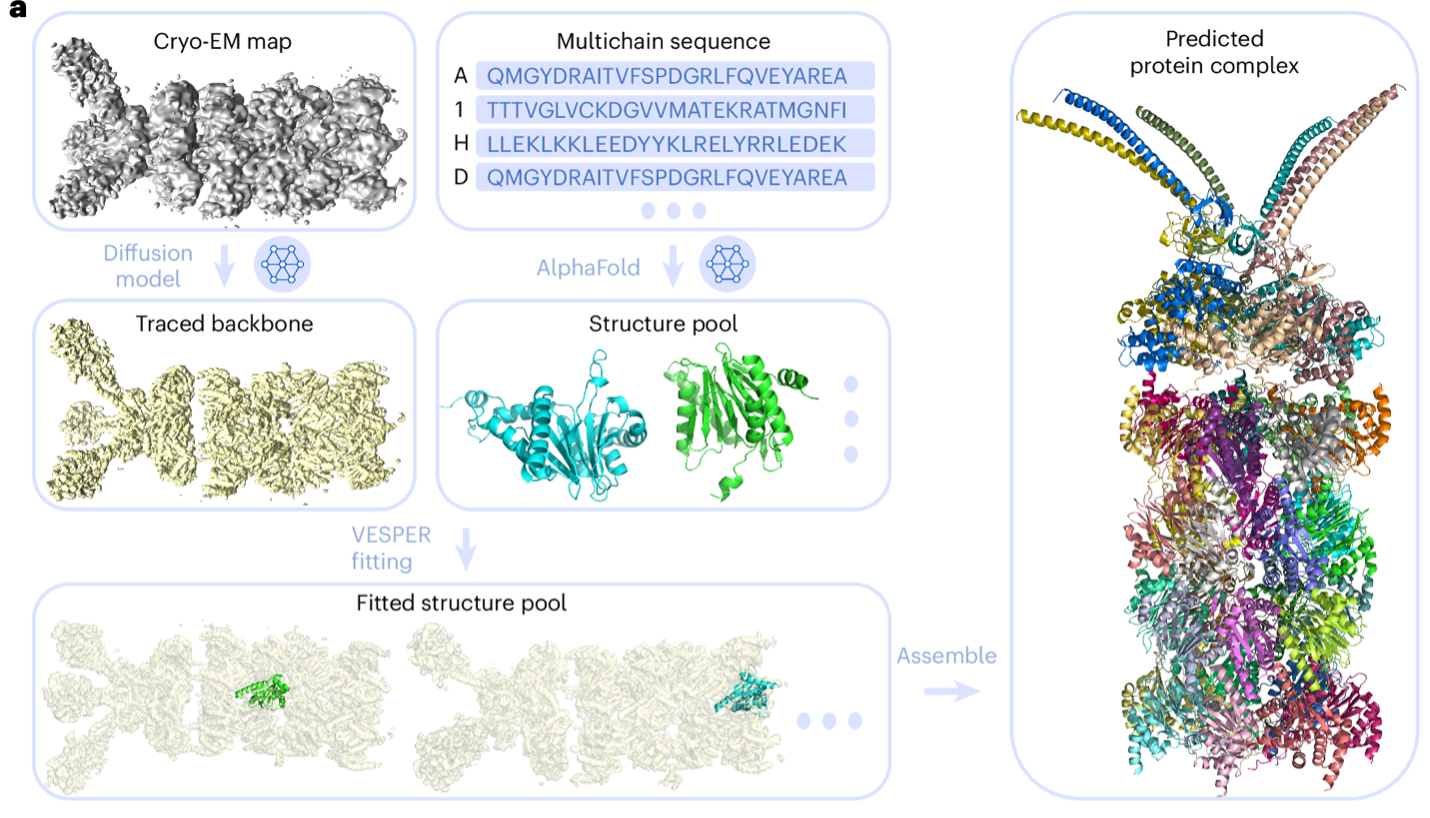
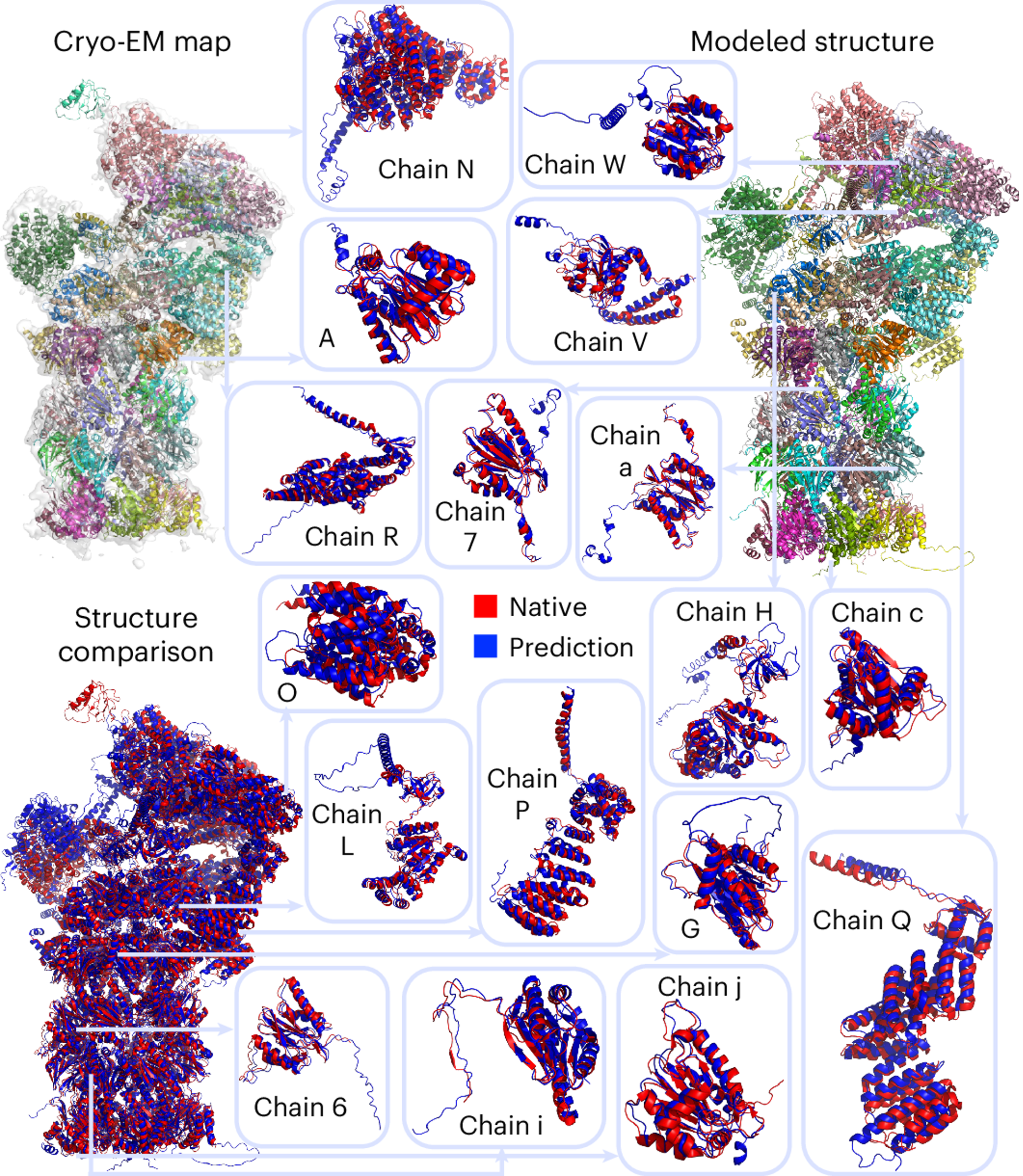
DiffModeler showed an average template modeling score of 0.88 and 0.91 for two datasets of 61/28 cryo-EM maps of 0–5 Å resolution and 0.92 for intermediate resolution maps (5–10 Å), substantially outperforming existing methodologies. Further benchmarking at low resolutions (10–20 Å) confirms its versatility, demonstrating plausible performance. This method can also be combined with our previous tool for nucleic acid structure modeling, CryoREAD, to model heterogeneous macromolecular structure. On the right, I show an example of a modeled protein complex with 47 chains, with a TM-Score of 0.94 (PDB ID: 6he9; EMDB ID: EMD-0213).
This work was motivated by my previous research experience with CryoREAD and DeepMainmast, where I discovered that de novo structure modeling is infeasible for low-resolution cryo-EM maps. However, such maps are widely available and significantly more affordable to obtain than high-resolution cryo-EM maps. During my review of structures solved by biologists, I noticed that manual structure fitting with AlphaFold2 or native single-chain structures was commonly employed to model protein complexes. This observation inspired me to develop an automated structure modeling tool specifically tailored for low-resolution maps. The outcome, DiffModeler, has been a significant success, with approximately 1,000 jobs submitted by biologists to our server for complex structure modeling. I am so happy to see the software being widely adopted and contributing meaningfully to the field of structural biology. We will remain committed to maintaining the server and its GitHub repository for the community and are excited about the potential for further structural discoveries enabled by this tool.
To help structural biologists to build protein complex from cryo-EM maps, the webserver is available at https://em.kiharalab.org/algorithm/DiffModeler, where users can simply upload the map and single-chain structures, then the structure will be modeled without installment. We also support sequence input for modeling, where the server will search PDB and AF2 database to obtain single-chain structure for modeling: https://em.kiharalab.org/algorithm/DiffModeler(seq). For macromolecular structure modeling with DNA/RNA, you can also use the service here: https://em.kiharalab.org/algorithm/ComplexModeler. Full source code is available in Github: https://github.com/kiharalab/DiffModeler. A detailed tutorial for DiffModeler usage is available at https://em.kiharalab.org/tutorial. If you have any questions or possible ideas to further improve DiffModeler, please contact Prof. Daisuke Kihara (dkihara@purdue.edu).
Follow the Topic
-
Nature Methods

This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Jul 28, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in