Discovery of a Potent Glutaminyl-peptide Cyclotransferase-like Protein (QPCTL) inhibitor with In Vivo Antitumor Efficacy
Published in Cancer
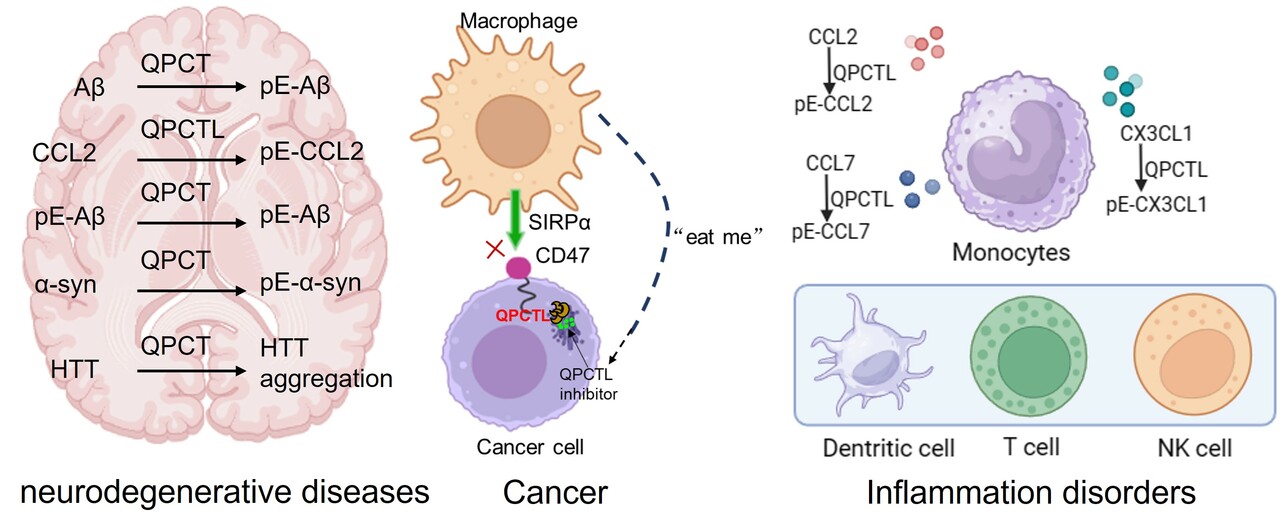
Cancer cells exploit immunosuppressive pathways to evade immune detection. Immune checkpoint therapy, which manipulates the immune system to attack tumors, has shown remarkable clinical success against various cancers. Integrin-associated protein (also known as Cluster of Differentiation 47, CD47), a crucial immune checkpoint molecule, helps cancer cells escape macrophage-mediated phagocytosis through interaction with SIRPα. CD47 is not only highly expressed on tumor cells, but also normal cells, such as red blood cells (RBCs). This poses a challenge in clinical trials, as some anti-CD47 antibodies may trigger unwanted side effects like anemia by promoting macrophage-mediated phagocytosis of RBCs. Therefore, there is an urgent clinical need to find new effective strategies for tumor immunotherapy. In 2019, Schumacher's group 1 and Wang's group 2 successively reported a key regulator, Glutaminyl-peptide cyclotransferase-like protein (QPCTL, also known as isoQC), that modulates CD47/SIRPα interaction. Genetic and pharmacological intervention of QPCTL attenuates the CD47/SIRPα interaction, leading to enhanced antibody-dependent cellular phagocytosis and cellular cytotoxicity of tumor cells. In 2022, Schumacher's group reported that QPCTL modulates the abundance of macrophages and monocytes as well as the expression of inflammatory factors in the tumor microenvironment 3. At the same period, Albert et al. 4 found that deletion of intracellular QPCTL limits chemokine function and remodels myeloid infiltration, thereby enhancing tumor immunity. Furthermore, Schloesser et al. 5 recently reported that the CD47-QPCTL axis is upregulated in senescent cells, leading to inhibition of macrophage-mediated clearance of apoptotic cells. These findings suggest that QPCTL is a promising therapeutic target in cancer immunotherapy.
Recently, Wang group and Xu group discovered a novel QPCTL inhibitor. This study provided a potent lead compound for drug development in tumor immunotherapy, and a highly active tool molecule for exploration of the biological functions of QPCTL in tumor immunity.
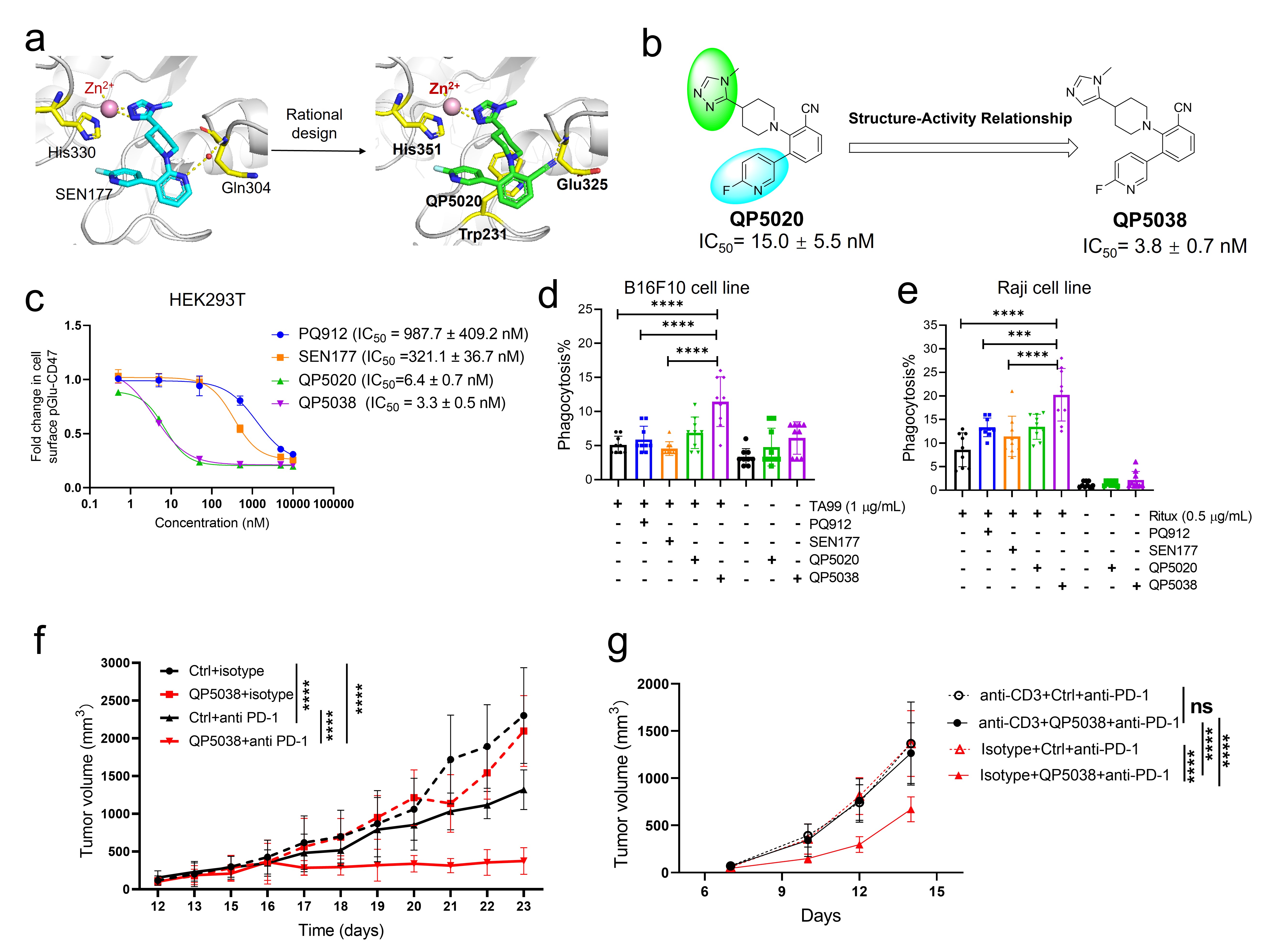
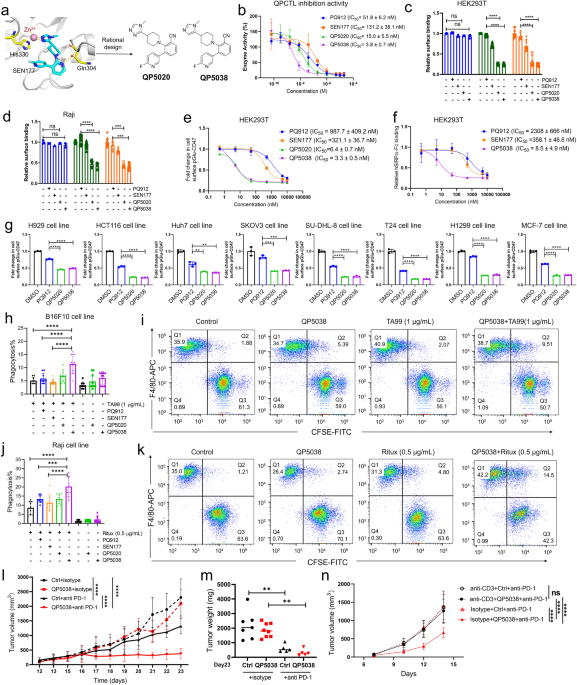
Figure 1 a. Docking analysis of SEN177 to QPCT and QP5020 to QPCTL. QPCT protein structure (left) (PDB: 6GBX) and QPCTL protein structure (right) (PDB: 3PB7) are downloaded from PDB protein structure database. b. Design of novel and potent QPCTL inhibitors. c. Dose-dependent inhibition of pGlu-CD47 following treatment with QPCTL inhibitors for 48 hours in HEK293T cells. d. Phagocytosis of control-treated (DMSO) (-) or QPCTL inhibitors-treated (+) B16F10 cells in the presence or absence of TA99 by mouse macrophages. e. Phagocytosis of control-treated (DMSO) (-) or QPCTL inhibitors-treated (+) Raji cells in the presence or absence of rituximab (Ritux) by mouse macrophages. f. Anti-tumor efficacy of QP5038 in mice. g. Anti-tumor efficacy of QP5038 in the presence or absence of the T cell depletion antibody.
Research on QPCTL inhibitors for tumor immunotherapy is still in its early stages, and only some small molecule inhibitors targeting QPCT, including PBD150, PQ912 and SEN177, exhibited inhibitory activity against QPCTL 6. Herein, we report the discovery of a series of novel QPCTL inhibitors using a combination of bioisostere-based and structure-based approaches. Initially, we analyzed existing data on protein X-ray structure and known inhibitors, paying particular interest to QPCTL inhibitor SEN177. The structure of human QPCT-SEN177 complex has been documented (Figure 1a). Since the active protein pocket of QPCTL shares a high degree of conservation with the QPCT structure, we docked SEN177 with the QPCTL structure. In this binding model, the triazole moiety of SEN177 binds the Zn ion of QPCTL in active pocket, the fluoro-pyridine interacts with His351, and pyridine forms a hydrogen bond with Glu325 mediated by a water molecule. Given that displacement of structural water molecule by a nitrile group may lead to significant enhanced affinity, we rationally replaced the nitrogen atom in pyridine moiety of SEN177 with a nitrile group and synthesized compound QP5020, which exhibited potent inhibition with IC50 of 15.0 nM (Figure 1a). We further focused on the triazole moiety and the fluoro-pyridine group and explored the structure-activity relationships (Figure 1b). Among them, QP5038 demonstrated the most potent inhibition of QPCTL with IC50 value of 3.8 nM, which is about 35-fold more potent than reported QPCTL inhibitors SEN177. This compound also exhibited a comparable inhibition to QPCT.
To investigate the effects of our QPCTL inhibitors on CD47 pyroglutamation and the ability to promote the macrophage-mediated phagocytosis of cancer cells, we treated HEK293T and cancer cells with QP5038 in vitro. QP5038 exhibited dose-dependent inhibition of pGlu-CD47 levels in HEK293T cells with remarkable IC50 values of 3.3 ± 0.5 nM (Figure 1c). Further, in vitro phagocytosis assay showed that QP5038 significantly boosted the phagocytosis of B16F10 cells in combination with TA99 treatment (Figure 1d) or Raji cells synergized with rituximab (Figure 1e). The efficiency of QP5038 on phagocytosis was much better than that of SEN177 and PQ912. Further, we assessed the potential therapeutic effects of QP5038 in combination with PD-1 inhibition in mice. Our data showed that the combination treatment of QP5038 with anti-PD-1 antibody dramatically suppressed both tumor growth and tumor weight comparing to each single treatment (Figure 1f). Moreover, depletion of T cell in mice using anti-CD3 antibody blocked the enhancement of QP5038 on anti-PD-1 antibody-mediated tumor inhibition, suggesting that the anti-cancer effect of QP5038 was due to the activation of immune response (Figure 1g).
In summary, our research identified QP5038 as a novel QPCTL inhibitor and provided a potent lead compound for drug development in cancer immunotherapy. Given that QP5038 exhibited potent inhibitory activity against QPCT/L, it is also expected to explore efficacy in treating neurodegenerative disorders and inflammatory disorders in the near future.
Corresponding author: Ping Wang, Professor, Tongji University. Wang's group focuses on tumor microenvironmental regulation and targeting. The long-term goal of the group is to discover new tumor therapeutic targets and develop new intervention strategies by revealing the mechanisms of tumor microenvironmental regulation.
Corresponding author: Shilin Xu, Principal Investigator, Shanghai Institute of Materia Medica, Chinese Academy of Sciences. The research interests of Xu’s group are structure-based design and synthesis of small molecule targeting epigenetic and immune targets, and discovery of tool molecules for new drug targets and mechanism of action studies.
Reference
- Logtenberg, M. E. W. et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat. Med. 25, 612-619, (2019).
- Wu, Z. et al. Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRPalpha-CD47 axis. Cell Res. 29, 502-505, (2019).
- Bresser, K. et al. QPCTL regulates macrophage and monocyte abundance and inflammatory signatures in the tumor microenvironment. Oncoimmunology 11, 2049486, (2022).
- Barreira da Silva, R. et al. Loss of the intracellular enzyme QPCTL limits chemokine function and reshapes myeloid infiltration to augment tumor immunity. Nat Immunol 23, 568-580, (2022).
- Schloesser, D. et al. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis. J Cell Biol 222, e202207097, (2023).
- Coimbra, J. R. M., Moreira, P. I., Santos, A. E. & Salvador, J. A. R. Therapeutic potential of glutaminyl cyclases: Current status and emerging trends. Drug Discovery Today 28, 103644, (2023).
Follow the Topic
-
Signal Transduction and Targeted Therapy
This is an international, peer-reviewed, open-access journal publishing articles related to signal transduction in physiological and pathological processes, alongside signal transduction-targeted therapeutics in the form of biological agents and small molecular drugs used to treat human diseases.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in