Engineering CRISPR base editors towards a permanent treatment for SMA
Published in Biomedical Research
For several years, I have been deeply interested in developing novel therapies for neuromuscular diseases. Spinal muscular atrophy (SMA) is still the leading genetic cause of infantile death worldwide and, although the success of approved therapies for SMA has minimized the high mortality rate early in life, current treatments have limitations and are not a permanent cure.
Since SMA is caused by mutations in the SMN1 gene, genome editing technologies that target the underlying genetic mutations in SMN1 could offer hope for a permanent cure for SMA. However, the heterogeneity and complexity of these mutations would require patient-specific therapeutics (which imposes challenges related to optimization and scaling thousands of editing approaches). A paralogous gene SMN2 is a main genetic modifier of SMA severity, where SMA subjects carrying multiple copies of SMN2 can partially compensate for the loss of SMN protein resulting from SMN1 loss-of-function. However, SMN2 produces only low levels of SMN protein compared to SMN1 due to a natural C-to-T substitution in exon 7 (“C6T”) that alters splicing and causes premature degradation of SMN.
In our recent study, we took advantage of the biology of SMN1 and SMN2 to develop a genome editing approach to restore SMN expression by targeting and correcting the SMN2 exon 7 coding sequence to an SMN1-like state (Figure 1a), towards the development of a pan-SMN1 mutation therapy for all SMA patients.
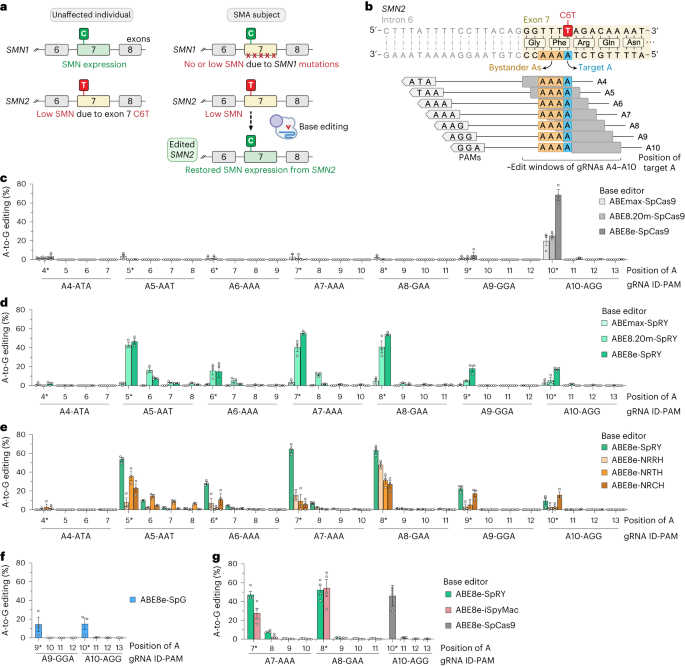
 Schematic of SMN1 and SMN2 in unaffected individuals and spinal muscular atrophy (SMA) patients. (b) Schematic of the SMN2 exon 7 C-to-T (C6T) polymorphism compared to SMN1, with base editor guide RNA (gRNA) target sites, their estimated edit windows, and their protospacer-adjacent motifs (PAMs).")
To develop a base editing approach to correct SMN2 C6T, we engineered and tested the efficiency of several CRISPR base editors capable of catalyzing A•T-to-G•C edits1-3. Importantly, the deaminase domain of these base editors can act in a narrow ~7-8 nucleotides ‘edit window’ within the target site at a fixed distance from the protospacer-adjacent motif (PAM; Figure 1b). Thus, the efficiency of base editing is dependent on the availability of Cas9 variant enzymes that can recognize PAMs at the appropriate distance upstream, towards maximizing editing efficiency of the intended base while minimizing unwanted editing of nearby bases that also fall within the edit window (bystander edits). In our study, we tested >100 combinations of guide RNAs (gRNAs) and adenosine base editors (ABEs) to understand the best combination to correct SMN2 C6T. One optimized combination that utilized a near-PAMless CRISPR-Cas enzyme, named SpRY4, resulted in up to 99% intended editing in SMA patient-derived fibroblasts (Figure 2a) with no bystander editing. In addition, we found that the DNA off-target profile of our optimized ABE-mediated C6T edit is highly specific in SMA-fibroblasts. As a result of this edit, we observed recued SMN protein levels to normal levels (Figure 2b). These results demonstrate that we could achieve high levels of SMN2 correction by optimizing and testing various ABE & gRNA combinations, leading to a durable edit that can restore SMN protein expression to near wild-type levels.
 A-to-G editing of the C6T opposite strand adenine in SMN2 exon 7 across five SMA fibroblast cell lines transfected with ABE8e-SpRY and gRNA A8. (b) SMN protein levels across three edited SMA fibroblast lines.")
Next, given that SMA is a neuromuscular disease primarily affecting the CNS (motor neurons), but also other peripheral tissues, we explored whether our editing approach could be translated into an in vivo model of SMA. Delivery of our optimized ABE and gRNA via dual adeno-associated virus (AAV) vectors into an SMA mouse model (Figure 3a) resulted in precise SMN2 C6T editing in bulk tissue. Notably, the levels of SMN2 C6T correction were sufficient to promote a significant 2- to 4-fold increase in SMN transcript levels in various tissues from SMA mice (Figure 3b). These editing levels led to a modest phenotypic change, which is expected due to the severity of this mouse model with a short period of intervention. To determine whether longer follow-up could result in higher SMN2 C6T editing, we analyzed a cohort of non-SMA SMND7 mice at 12 weeks after a P1 ICV injection and observed up to 20% on-target editing in the brain and up to 30% editing in liver (Figure 3c-d). Thus, other approaches able to provide an extended therapeutic window in this severe SMA mouse model are likely necessary for a better phenotypic rescue before irreversible pathology. Another recent contemporary study performed an experiment involving co-administration of an antisense oligonucleotide treatment (i.e., nusinersen) combined with a similar in vivo ABE-mediated SMN2 C6T editing strategy5. Nusinersen extended the therapeutic window of SMA mice sufficiently to permit superior phenotypic improvements from C6T editing in this severe SMA mouse model. Importantly, we observed comparable levels of on-target editing when comparing our C6T base editing approach to the base editor from this independent study5, suggesting that different customized base editors can be applied to provide durable benefits for SMA patients via strategies that are agnostic of their SMN1 mutation.
 Schematic of P1 ICV injections in SMND7 mice with dual AAV9 vectors that express intein-split ABE8e-SpRY and gRNA-A8. (b) SMN exon 7 mRNA expression. (c) Schematic of P1 ICV injections in SMND7 mice with long-term follow up. (d) A-to-G editing of SMN2 exon 7 adenines following ICV injections of AAV encoding ABE8e-SpRY with gRNA A8.")
Our study is one of the first demonstrations of optimizing and utilizing customized base editors for SMA and, more broadly, establishes a proof-of-concept of a PAM-flexible CRISPR-Cas enzyme (SpRY) that can be applicable to treat various disease-causing mutations. This blueprint to rapidly screen editing strategies with a single versatile enzyme in patient-derived cells, followed by translatability into in vivo models, could expedite the development of new genetic medicines. Some key aspects of our study may be extensible to the treatment of other diseases, such as the use of genome editing to modify a cognate paralog gene instead of editing patient-specific mutations, and the utility of screening gRNAs to enable precise on-target editing with low (if any) bystander editing for sites with poly-adenine stretches. Thus, the flexibility of engineered enzymes enables genome editing to achieve highly precise edits, and we envision that the application of these novel enzymes to other severe genetic diseases will motivate and enable the development of new therapies.
In summary, the continued development of our base editing approach should provide long-lasting genetic treatment for SMA, where a durable one-time edit offers major advantages compared to existing therapies. Our work highlights the potential of customized base editors to install corrective genetic edits efficiently and safely, which can be extended to a wide range of human diseases.
References
1 Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol 38, 892-900 (2020). https://doi.org/10.1038/s41587-020-0491-6
2 Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464-471 (2017). https://doi.org/10.1038/nature24644
3 Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 38, 883-891 (2020). https://doi.org/10.1038/s41587-020-0453-z
4 Walton, R. T., Hsu, J. Y., Joung, J. K. & Kleinstiver, B. P. Scalable characterization of the PAM requirements of CRISPR-Cas enzymes using HT-PAMDA. Nat Protoc 16, 1511-1547 (2021). https://doi.org/10.1038/s41596-020-00465-2
5 Arbab, M. et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science 380, eadg6518 (2023). https://doi.org/10.1126/science.adg6518
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Microphysiological systems for advanced modeling, high-throughput evaluation, and clinical translation
Publishing Model: Hybrid
Deadline: Dec 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in