Experiments of nature: Using CRISPR to harness the power of natural variants
Published in Bioengineering & Biotechnology, Microbiology, and Cell & Molecular Biology
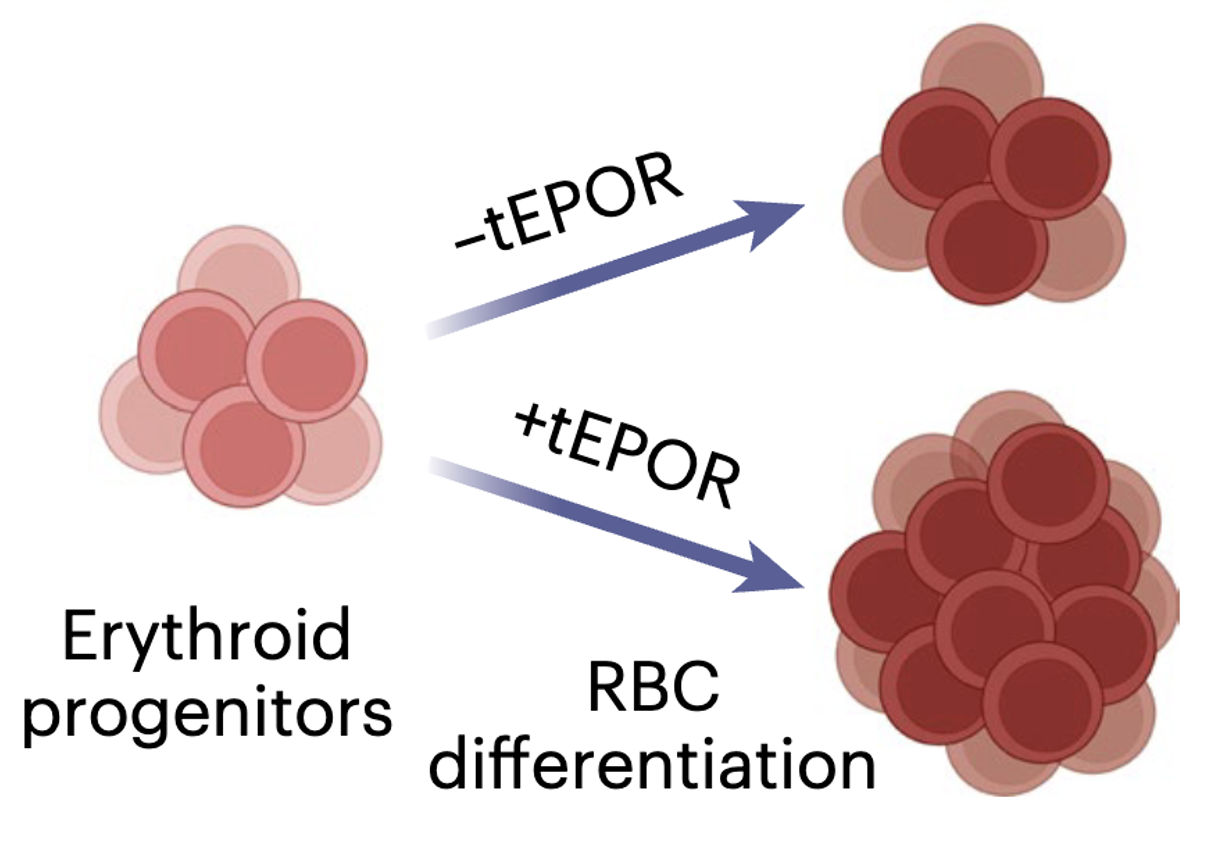
In December 2023, the FDA approved the very first genome editing therapy—providing a potentially lifelong therapy for sickle cell disease1. This was a landmark moment for the genome editing field and will be the first among a wave of CRISPR-based therapies for genetic diseases.
To those in the field, it came as no surprise that the first FDA-approved CRISPR-based genome editing therapy targeted sickle cell disease. This disorder has long been considered a low-hanging fruit for precision medicine. For one, the genetics of sickle cell disease could not be simpler. While many disorders, referred to as monogenic diseases, are caused by various mutations within the same gene, sickle cell disease stands out. Not only do patients with sickle cell disease have mutations in the same gene, beta-globin, but remarkably, they share the exact same genetic change: a single A-to-T substitution. This is especially remarkable given the vast 6-billion base-pair expanse that comprises the human diploid genome. In addition, sickle cell disease is quite common for such a devastating disease, with millions of severely affected patients worldwide. Since there has always been heavy selection pressure against pathogenic alleles, it is reasonable to ask then how such a devastating mutation became so prevalent?
Most genetic variation arises spontaneously. The environment then dictates which variants are likely to be passed to future generations and which are not. This is the mechanism of evolution and adaptation, in what can be referred to as “experiments of nature”. In this natural experiment, you would expect that deleterious variants like the sickle mutation would reduce fitness and rapidly disappear from the population. However, the prevalence of sickle cell disease tells a different story. While those with homozygous mutations have a terrible disorder, heterozygous carriers do not. In fact, carriers are resistant to malaria, imparting a major survival advantage to those with this variant (Figure 1A). Based on the wide prevalence of the sickle mutation, we can conclude that the survival advantage imparted to heterozygous carriers must far outweigh the survival disadvantage of homozygotes (Figure 1B). Human populations felt the heavy selection pressures of malaria, and nature neatly crafted a single-variant solution.
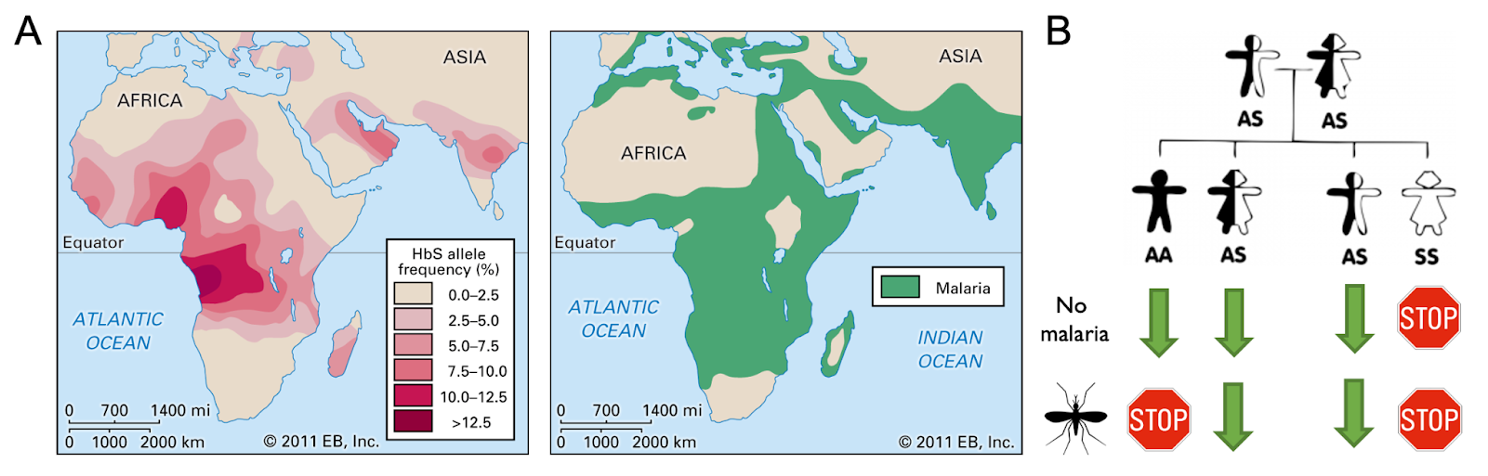
Figure 1: A) Map showing regional sickle cell disease (HbS) allele frequencies (left panel); map showing regions most heavily impacted by malaria (right panel; adapted from Encyclopedia Britannica). B) Example of genetic outcomes resulting from two sickle cell disease carriers. Expected selection pressures applied in the context of regions with and without malaria. Stop sign indicates selective disadvantage; green arrow represents selective advantage.
In contrast to carriers, the negative consequences for patients with homozygous sickle mutations appear to outweigh the benefits against malaria. However, any time there is substantial negative selection pressure against a large population, nature has an opportunity to run more experiments. Incredibly, there are now people with homozygous sickle mutations who do not suffer from the disease. Research into this rare population found that they have a benign disorder called “hereditary persistence of fetal hemoglobin”, where the precursor to beta-globin was never silenced due to mutations in normal regulatory machinery. These mutations allow fetal hemoglobin to remain expressed so that even people with mutations in beta-globin are able to effectively transport oxygen throughout their bloodstream. Because this experiment of nature was so successful at mitigating the detrimental effects of sickle cell disease, scientists deployed the same strategy in the first FDA-approved CRISPR-based gene therapy for sickle cell disease—using CRISPR to reactivate fetal hemoglobin in patients with sickle cell disease2. To date, over seventy patients have been effectively treated for their disease using this strategy.
This success story shows how genome editing tools can be used to recapitulate natural variants and solve clinical problems. And although there is an FDA-approved strategy to treat sickle cell disease, it is clear that additional hurdles exist that prevent patients from fully benefiting from this therapy. For one, the approved therapy comes with the requirement of chemotherapy to clear space in the bone marrow for a patient’s edited hematopoietic stem cells (HSCs). Once there, patient HSCs behave normally and produce the many cell types of the immune and blood systems. However, the most important cell type for correction of sickle cell disease is the red blood cell (RBC). This creates the need for a relatively high percentage of edited patient HSCs to engraft in the bone marrow in order to reach a curative threshold. However, if there was some way to increase production of RBCs from patient HSCs, then a lower dose of HSCs could still be clinically meaningful. This may then enable clinicians to reduce or eliminate the chemotherapy regimen that stands as a major barrier to safe translation of the CRISPR therapy.
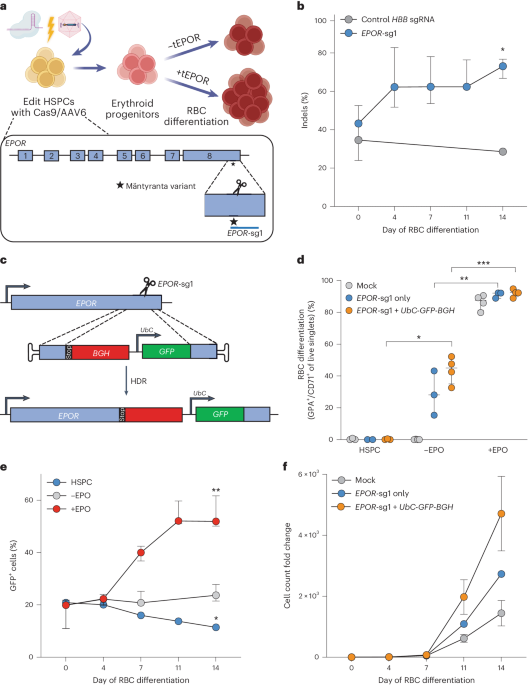
To pursue this idea, we looked for examples in nature, as other scientists have similarly done in the past, that could be deployed using genome editing tools. We found a disorder called “congenital erythrocytosis” where people have higher than normal RBC levels and consequently elevated hemoglobin. The genetic root of this phenotype was identified by studying the family of a Finnish Olympic gold medal-winning cross-country skier. What began as a blood doping accusation, ended as the discovery that this Finnish skier had a mutation in his erythropoietin receptor (EPOR) which made his body hypersensitive to EPO3. In this cohort, these mutations imparted increased fitness with few discernible disadvantages; an experiment of nature that increased the likelihood that this variant would be passed to subsequent generations.
While sickle cell disease is not particularly prevalent in Finland, genome editing tools give us the ability to take naturally occurring variants in one population and apply them to another. Therefore, in our study we leveraged CRISPR tools to introduce naturally occurring EPOR mutations and found this significantly increased RBC output from edited HSCs. We then showed that this selective advantage could be coupled to a therapeutic edit in HSCs from patients with sickle cell disease to increase production of corrected RBCs. We believe this work not only has the potential to reduce barriers to safe and effective cures for sickle cell disease, but also showcases how experiments of nature can provide the basis for novel genome editing strategies. As genome sequencing becomes more commonplace and clinically routine, it is likely that we will discover and characterize many more experiments of nature that could be repurposed for the benefit of patients.
References
- Harris, E. Sickle cell disease approvals include first CRISPR gene editing therapy. JAMA 331, 280 (2024).
- Frangoul, H. et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
- de la Chapelle, A., Träskelin, A. L. & Juvonen, E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci USA 90, 4495–4499 (1993).
Follow the Topic
-
Nature Biomedical Engineering

This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.
Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in