From Genome to Transcriptome: Charting the Yeast Genetic and Environmental Perturbation Map
Published in Cell & Molecular Biology and Genetics & Genomics

What is Heterogeneity?
The first time you look at cells from any organism under the microscope, you realize that even clonal and therefore genetically identical cells look different. Most likely you have pictures of really odd or funny-looking cells, at least we do. These differences in shape, morphology, and size also translate to more complex traits and influence essential processes like growth rate, stress resistance or sensitivity, and ultimately survival. While this heterogeneity has been known for a long time and is readily observable at first glance, the molecular underpinnings remained unexplored until the era of single-cell which provided an unbiased quantitation of heterogeneity.
Since its development, single-cell RNA-seq (scRNA-seq) has served to catalog and describe the variance within populations across multiple scales: within cell types, tissues, organisms, disease states, and recently in vivo. However, understanding causal relationships required the incorporation of genetic perturbations into these systems. The combination of single-cell genetic perturbations with single-cell RNA-seq (Perturb-seq) has revolutionized our understanding of cellular behavior by allowing us to dissect the relationships between genotype, gene expression, and phenotype at unprecedented resolution. Perturb-seq was pioneered in mammalian cells through CRISPR-based genetic perturbations, and in recent years we have seen how the scale of measured perturbations has exponentially increased. However, few studies have reached genome-wide scale, and even fewer organism-level studies, despite this rapidly becoming a powerful reality. The field is ready to move beyond associative and correlative findings towards a causal understanding of biological systems.
The Yeast Approach
However, pooled CRISPR screens are often assayed out of context in vitro and transfected with CRISPR libraries. Although they are highly advantageous as they enable large-scale genetic perturbations, they lead to different perturbations for each gene. The perturbations are often not isolatable from the pool. We found that yeast offered a unique framework as unicellular clonal populations with very few intrinsic sources of heterogeneity. The application of single-cell technologies in yeast has lagged behind due to technical challenges posed by a small transcriptome and the presence of a cell wall. In recent years, several scRNA-seq methods have emerged, reducing the technical gap.
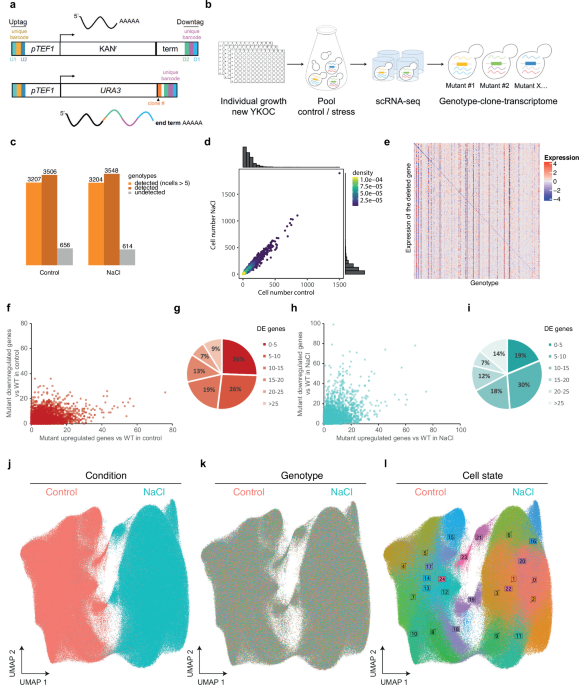
For decades, the versatile yeast genome has facilitated the development of revolutionary tools for functional genomics. These tools include various yeast collections, such as the Yeast Knock Out Collection (YKOC), which enables the deletion of non-essential genes, and other collections allowing genome-wide protein localization studies. However, the application of genome-scale Perturb-seq remained elusive due to the lack of an integrated system to trace genomic perturbations at the single-cell level combined with scRNA-seq. Building on the concept of expressible barcodes, we reconfigured the yeast YKOC. By generating RNA-traceable mutants, we could link each cell's genotype and transcriptional phenotype for each genetic perturbation in control and stress conditions. This has allowed us to build a high-resolution single-cell transcriptome atlas by profiling 3,500 yeast mutants.
Key Findings
Cell state map: We uncovered that transcriptional heterogeneity reflects a myriad of cell states within the yeast population, each characterized by specific transcriptional architectures. These states were influenced by both intrinsic factors (genetic makeup) and extrinsic factors (environmental conditions).
State Attractors: Certain mutants act as "state attractors," meaning they could influence the occupancy and transition of cell states within the population. This modulation of cell states resulted in differential fitness, highlighting the importance of transcriptional heterogeneity in adaptation to stress.
Impact on Fitness: We found that mutants with decreased transcriptional heterogeneity under control conditions exhibited significantly higher fitness when exposed to stress. Conversely, those with high transcriptional heterogeneity tended to show lower fitness in stressful environments. This suggests that while some variability can be beneficial, excessive heterogeneity may lead to vulnerabilities.
The Role of Environmental Stress
Environmental stressors, such as changes in temperature, nutrient availability, or exposure to toxins, can significantly impact gene expression and cellular behavior. Our study highlighted that yeast cells respond to stress by altering their transcriptional states, which can lead to different survival strategies. Interestingly, most of the cell states we identified were conserved across conditions suggesting these are robust to genetic and environmental perturbations. For example, under osmotic stress, certain gene expression patterns were activated to help the cells cope with the adverse conditions.
Why does this matter?
Our dataset provides a comprehensive look at genotype-to-transcriptome relationships at a single-cell resolution. By leveraging advanced genetic tools and single-cell transcriptomics, we have mapped the intricate landscape of gene expression and its impact on cellular behavior. In summary, transcriptional heterogeneity is not just a biological curiosity; it is a fundamental aspect of how cells navigate their environments, adapt to challenges, and ensure their survival.
Please find out more by reading our latest paper in Nature Communications.
Authors
Written by Mariona Nadal-Ribelles, Carme Solé, Yaima Matas, Eulàlia de Nadal and Francesc Posas
Poster image was generated in part by chat GPT.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in