How AI-Generated Binding Conformations Could Improve Drug-Protein Binding Affinity Predictions? Meet the Folding-Docking-Affinity Framework
Published in Chemistry, Cell & Molecular Biology, and Computational Sciences
Predicting how strongly a drug binds to a protein—a concept known as binding affinity—is a key step in discovering new medicine. Ideally, we want to make these predictions both quickly and accurately, especially during the early stages of drug development. But that’s no easy task.
At the heart of binding affinity lies the physical interaction between atoms in the protein and the drug. Traditional methods often rely on force fields governed by physical principles to model these atom-level interactions. So, it’s natural to think that starting from the atomic structure should help us understand how a drug and a protein interact.
When a protein undergoes changes, such as mutations or chemical modifications, its structure can shift, which may influence how a ligand binds to it. In theory, if we can figure out the 3D shape of the protein-drug complex, we’re one step closer to knowing how tightly they’ll bind.
However, most existing deep learning methods for binding affinity prediction ignore the binding pose, meaning they don’t model how the drug physically fits into the protein. While these “docking-free” models are easy to train end-to-end, they often miss important atom-level interactions, making their predictions harder to interpret. Can we improve on this?
The Challenge: Where Do We Get These 3D Structures?
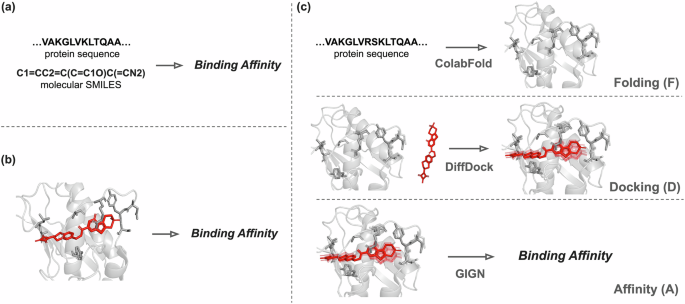
Recent breakthroughs in deep learning have made it easier to predict 3D protein structures (like with AlphaFold) and to simulate how drugs might dock to them (using tools like DiffDock). However, these advances haven’t yet cascaded down to the task of predicting binding affinity. We saw an opportunity here.
We introduce the Folding-Docking-Affinity (FDA) framework— that (1) folds the protein into its 3D shape, (2) docks the drug to the protein, and (3) predicts the binding affinity based on the computed binding structure.
Why This Framework Matters
There’s a big data imbalance in the field. We have far more binding affinity data than high-quality protein-drug structural data. Our end-to-end FDA framework bridges this gap by leveraging a limited set of structural data to build structure-aware models for large-scale binding affinity prediction. What's more, because it’s end-to-end, the structural prediction modules can be improved over time through feedback from the affinity predictions.
In the first version of FDA, we kept the folding and docking models parameters fixed and trained only the final affinity prediction model using data from benchmark datasets like DAVIS and KIBA. The results? FDA performed on par with leading docking-free methods. Interestingly, it performed even better in the more difficult case of predicting for unseen proteins and drugs, suggesting the framework’s potential.
What Happens When We Use Real vs. Predicted Structures?
We did an ablation study to test how different types of structural inputs affect predictions. Surprisingly, the fully AI-generated structures performed better than those based on real crystal structures. This was unexpected—after all, AI-generated structures introduce noise.
We think this noise might help. It may teach the model to generalize better by learning a smoother "landscape" of how binding affinity changes with structures. In other words, a little uncertainty might make the model more flexible and robust.
Putting the Hypothesis to the Test
To explore this idea, we trained the model using not just one predicted binding pose per protein-ligand pair, but several. The result? The model’s performance improved, even surpassing state-of-the-art docking-free methods. This suggests that introducing multiple slightly different binding poses can be a powerful form of data augmentation.
Where Do We Go from Here?
By incorporating 3D structural context—even when derived from AI models—we take a meaningful step toward bridging the gap between molecular structure prediction and drug-protein affinity modeling. We envision that the FDA framework can serve as a foundation for future studies aiming to integrate protein structure, binding pose, and affinity prediction into a unified and interpretable framework.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in