IL-33 enhances Jagged1 mediated NOTCH1 intracellular domain (NICD) deubiquitination and pathological angiogenesis in proliferative retinopathy
Published in Healthcare & Nursing
The retina consumes a high proportion of oxygen, and polyunsaturated fatty acids and is susceptible to oxidative stress. The retinal and choroidal vascular plexi regulate the elevated metabolic supply to the retina, and any abnormality or breakdown in these vascular plexi leads to tissue ischemia and pathological retinal angiogenesis. Current treatment regimens for these neovascular diseases, such as laser photocoagulation and anti-VEGF therapies are effective but carry risks. Laser photocoagulation therapies lead to the development of macular edema, visual field loss, and reduced night vision. Anti-VEGF therapies require regular intravitreal injections, leading to infections and endophthalmitis. In addition, anti-VEGF therapies lead to tractional retinal detachment and retinal photoreceptors atrophy. Recent studies have shown that NV is governed by a complex interplay between immune cells and inflammatory cytokines. Furthermore, it has also been reported that immune cells are recruited to the hypoxic tissue to induce the expression of cytokines to make a pro-angiogenic microenvironment. The present understanding of the role of cytokines and inflammatory processes in the pathogenesis of retinal NV is at an early stage, and the mechanism(s) by which inflammatory processes regulate visual dysfunction needs to be addressed.
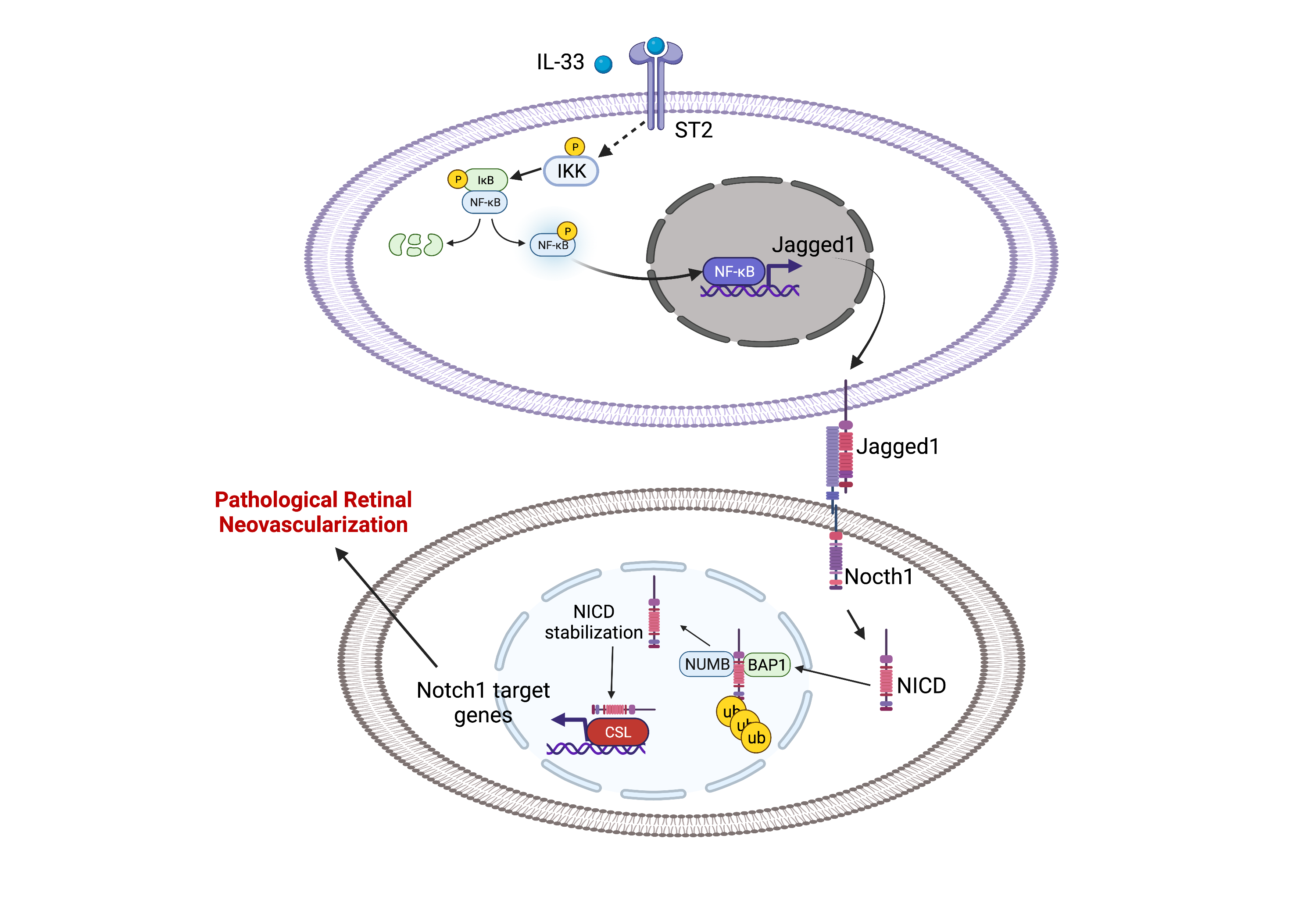
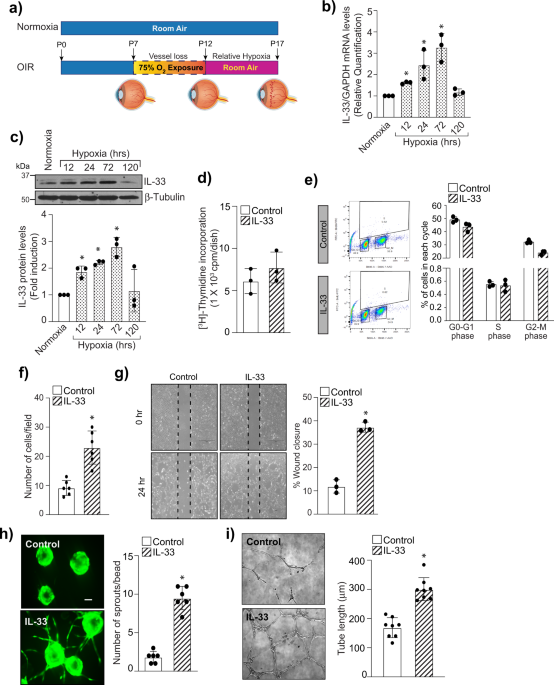
The role of IL-33 in angiogenesis is still not delineated, with the majority of reports showing a proangiogenic role for IL-33, while a few demonstrate its anti-angiogenic role. We observed that genetic deletion or siRNA-mediated downregulation of IL-33 reduces pathological NV in a murine model of oxygen-induced retinopathy (OIR) with no effect on the normal retinal repair. Furthermore, their fluorescent activated cell sorting (FACS) data revealed that the increase in IL-33 expression was mostly found in endothelial cells (ECs) of the hypoxic retina and conditional genetic deletion of IL-33 in retinal ECs reduces aberrant angiogenesis observed in ischemic retinopathies. Here we identified the functional role of IL-33 induced Jagged1/Notch1 signaling in post-ischemic neo-angiogenesis. In vitro studies using human retinal microvascular endothelial cells (HRMVECs) show that IL-33 induces sprouting angiogenesis and requires NFkappaB-mediated Jagged1 expression and Notch1 activation. Our data also suggest that IL-33 enhances de-ubiquitination and stabilization of Notch1 intracellular domain via its interaction with BRCA1-associated protein 1 (BAP1) and Numb in HRMVECs and a murine model of oxygen-induced retinopathy.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in