DNA methylation detection using IMPRESS as a novel strategy for multi-cancer diagnosis
Published in Genetics & Genomics and Biomedical Research
Cancer remains one of the deadliest diseases globally, with common types like breast, lung, colorectal, and prostate cancer each exceeding 1.4 million cases annually. Diagnoses often occur in advanced stages due to the lack of symptoms and effective screening programs, highlighting the need for reliable diagnostic biomarkers. DNA methylation is a promising biomarker for cancer detection, as tumors exhibit distinct methylation patterns early in carcinogenesis. While numerous studies have explored the methylome, few successful methylation biomarkers have been implemented clinically.
Current DNA methylation analysis relies heavily on bisulfite sequencing, the gold standard, despite its limitations such as DNA degradation. Alternative bisulfite-free techniques, such as affinity-based methods (e.g., MeDIP-seq), generally offer lower accuracy. Modern technologies like droplet digital PCR (ddPCR) increase sensitivity but cannot detect multiple targets simultaneously. Recent advancements include enzymatic methods like Enzymatic Methyl sequencing (EM-seq), which aim to reduce reliance on bisulfite conversion, but these methods are costly, hindering their routine diagnostic use. Up until now, there was a lack for a highly multiplexable, cost-effective and sensitive method for DNA methylation detection.
Combining a novel technology with a novel biomarker panel for multi-cancer detection
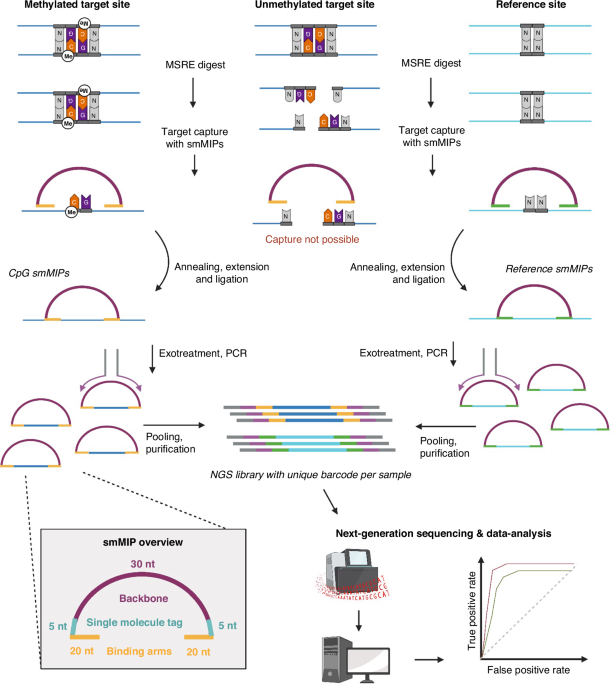
Our paper introduces a novel technique, IMPRESS (Improved Methylation Profiling using Restriction Enzymes and smMIP sequencing), combining methylation-sensitive restriction enzymes (MSREs) with Single-molecule Molecular Inversion Probes (smMIPs). MSREs have long been used for methylation analysis, while smMIPs are effective for capturing and enriching specific genomic regions, suitable for multiplex analysis of thousands of regions. Though smMIPs have been used in various research fields, our study pioneers their application in DNA methylation detection. The IMPRESS protocol involves digestion of DNA with MSREs, capturing methylated CpG regions using smMIPs, circularizing the DNA fragments, and amplifying them through PCR. Subsequently, sequencing is performed via Next Generation Sequencing.
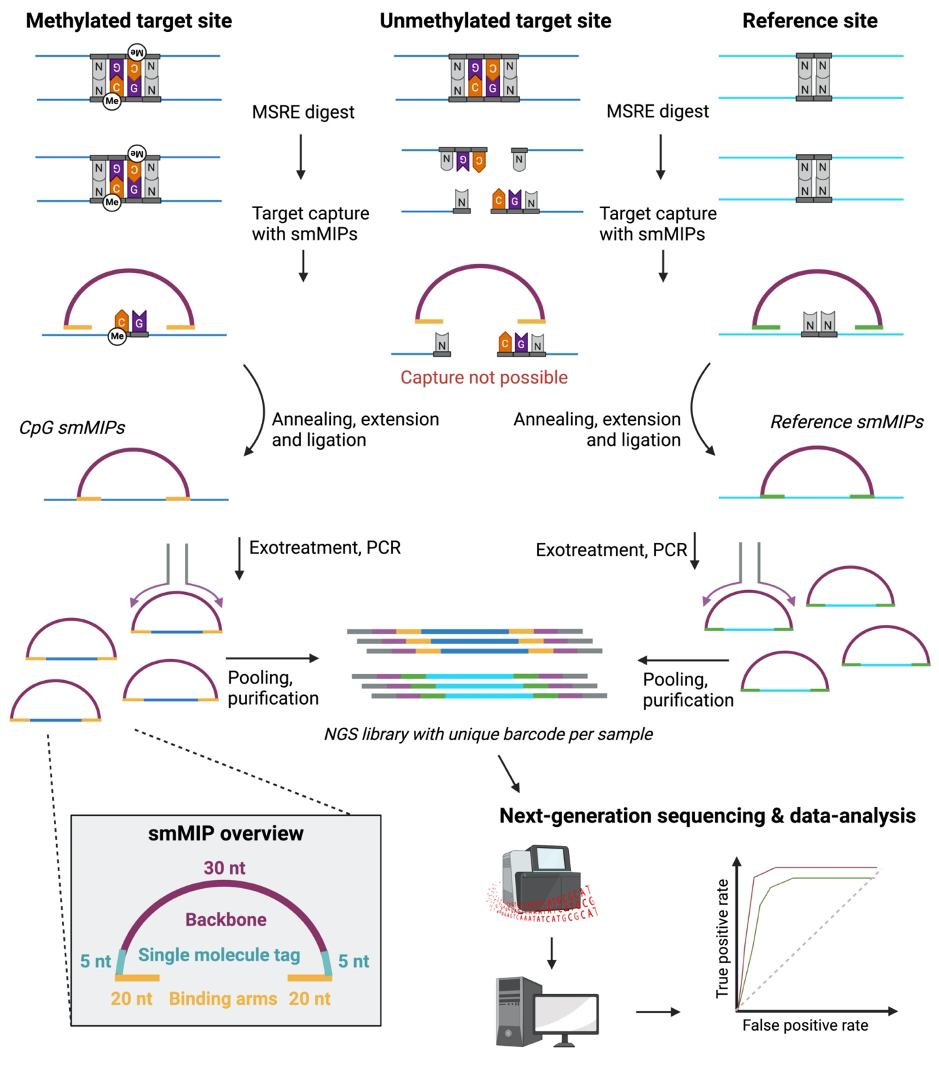
Besides the novel technology, we also developed a diagnostic biomarker assay to differentiate tumor samples from normal ones. Biomarker targets were selected based on 450K methylation data from tissue samples of eight cancer types and normal adjacent tissues, including normal blood samples, ensuring future applicability for plasma-derived liquid biopsies. We selected CpG sites that are consistently hypermethylated across all tumor types.
For data analysis, a bioinformatic pipeline was developed using Snakemake, handling outputs from MiSeq and NextSeq platforms, and involved steps like barcode splitting, quality trimming, mapping, duplicate removal, and read filtering. Classifier models based on NGS data were constructed and validated using linear discriminant analysis (LDA) and ROC curve analysis, optimizing for sensitivity and specificity.
The performance of IMPRESS
As an important first step, the efficiency of our four MSREs was tested on both methylated and unmethylated lambda DNA samples. The results showed that unmethylated samples were efficiently digested, with a ΔCt value of 17, indicating that approximately only 1 in 131,000 DNA molecules were not digested. In contrast, methylated samples had an average Ct value of 5.7, demonstrating that methylation effectively blocks digestion by the MSREs. To then assess the repeatability of the IMPRESS technique, two independent experiments were conducted on 29 fresh frozen tissue samples. The Pearson correlation coefficient of 0.99 indicated high repeatability.
The performance of the biomarker panel with IMPRESS
During development our multi-cancer biomarker panel, 1,791 hypermethylated CpG sites were selected from the methylation analysis of eight cancer types and healthy blood samples. Additionally, 600 reference sites without MSRE recognition site or withour CpG site were chosen to estimate the effective total amount of input DNA and allow normalization of results. For all selected target sites, smMIPs were designed.
To evaluate the biomarker panel in combination with the IMPRESS technique, a sequencing library was prepared from 111 fresh frozen tumor samples, 114 normal adjacent fresh frozen tissue samples, and 35 whole blood samples. The normalized counts of CpG smMIPs were used to differentiate between methylated (tumor) and unmethylated (normal) samples. The final dataset included counts of 2,312 CpG targeting smMIPs for 259 samples.
A classifier model was built using 358 single-tiled CpG smMIPs, achieving a sensitivity of 0.95, specificity of 0.91, and accuracy of 0.92. Validation with digital droplet PCR (ddPCR) confirmed that the IMPRESS technique performs at least as well as the gold standard ddPCR.
The application potential of IMPRESS in liquid biopsies
Further experiments tested the potential of IMPRESS for liquid biopsies, showing effective digestion and capture of cell-free DNA (cfDNA) with as little as 5ng input DNA. Furthermore, the limit of detection for methylation was found at 4.04%. While not yet tested on liquid biopsies from cancer patients, IMPRESS is showing promising preliminary results. With further optimization, this approach could enhance early cancer detection, offering a cost-effective, high-throughput alternative to existing methods.
Looking ahead: the future of IMPRESS
Besides its application in cancer, IMPRESS can become useful across various research fields. The IMPRESS technique enables targeted biomarker sequencing, offering significant advantages in multiplexing and cost-effectiveness compared to methods like ddPCR and genome-wide techniques. Our method allows the analysis of thousands of target sites at a high sample throughput, reducing sequencing costs and making it easily implementable in standard labs with straightforward protocols. It can potentially integrate different genetic analyses such as mutation and CNV analysis into one assay, moving towards a simple multi-omics strategy.
In conclusion, we developed a novel method for sensitive detection of DNA methylation, we developed a multi-cancer methylation biomarker panel, and we combined those two into a multi-cancer detection assay. The IMPRESS technique demonstrated high sensitivity and applicability for low amounts of fragmented DNA, crucial for liquid biopsies and early cancer detection. This novel approach offers a promising, cost-effective platform for the detection and follow-up of various diseases.
Follow the Topic
-
British Journal of Cancer
This journal is devoted to publishing cutting edge discovery, translational and clinical cancer research across the broad spectrum of oncology.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in