Molecular alignment as a shortcut to ultrafast-charging lithium metal batteries
Published in Chemistry

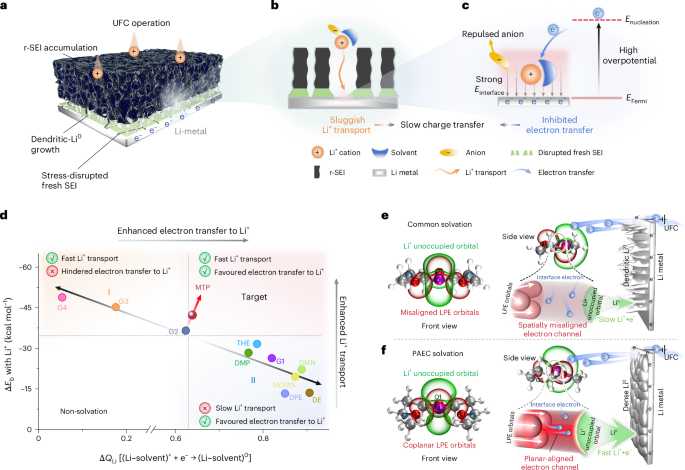
Fast charging has become one of the defining challenges in modern battery research. From electric vehicles to portable electronics, the expectation is clear: batteries should charge faster, last longer, and remain safe. Yet, for lithium metal batteries, despite their enormous promise, fast charging often exposes their most fundamental weaknesses.
When we started this work, we were repeatedly confronted with a familiar dilemma. Lithium metal offers unparalleled energy density, but when pushed to high charging rates, it tends to respond badly: large overpotentials, unstable lithium deposition, and rapid performance decay. Many excellent studies have focused on protecting lithium metal with artificial interphases or optimizing electrode architectures. However, we began to ask a different question: what if the bottleneck lies not only in the electrode, but in how the electrolyte itself mediates electron transfer at the molecular level?
Looking beyond ions
Electrolytes are usually discussed in terms of ionic conductivity, solvation strength, or electrochemical stability windows. Electrons, by contrast, are often treated as visitors that briefly cross the interface and immediately disappear into the metal. But during fast charging, electron transfer is no longer an instantaneous event, it becomes a rate-determining process.
This realization prompted us to look more carefully at the interfacial region, where solvent molecules, ions, and electrons coexist under extremely strong electric fields. Rather than viewing the electrolyte as a random molecular soup, we began to consider whether its molecular organization of the solvation could actively facilitate or hinder electron transfer.
An unexpected role of molecular alignment
What emerged from our investigation was a surprisingly simple yet powerful insight: with the appropriate molecular structure, solvent lone-pair electrons can adopt a planar alignment. Crucially, we discovered that this co-planar configuration is not limited to the static crystals we prepared; it persists as the preferred energetic state even within the dynamic liquid electrolyte and at the charged electrode-electrolyte interface. This alignment enhances solvation ability and establishes a continuous electronic pathway. These dynamically coupled lone pairs collectively create what we term 'molecularly planar-aligned electron-channels' (PAEC), offering a shortcut for efficient interfacial electron transfer. Rather than relying on rigid permanent structures, this alignment is sustained by subtle intermolecular interactions and the interfacial electric field.
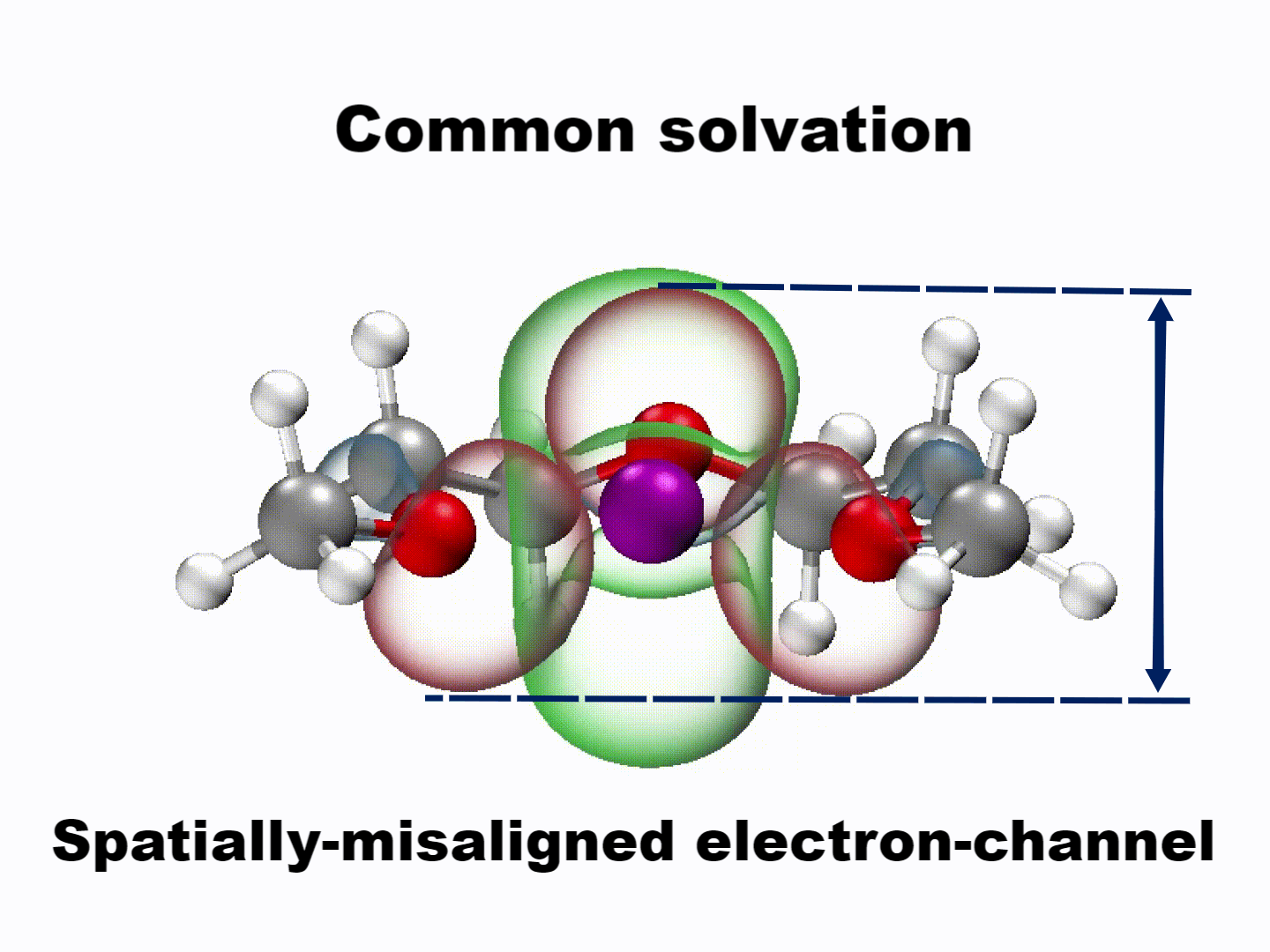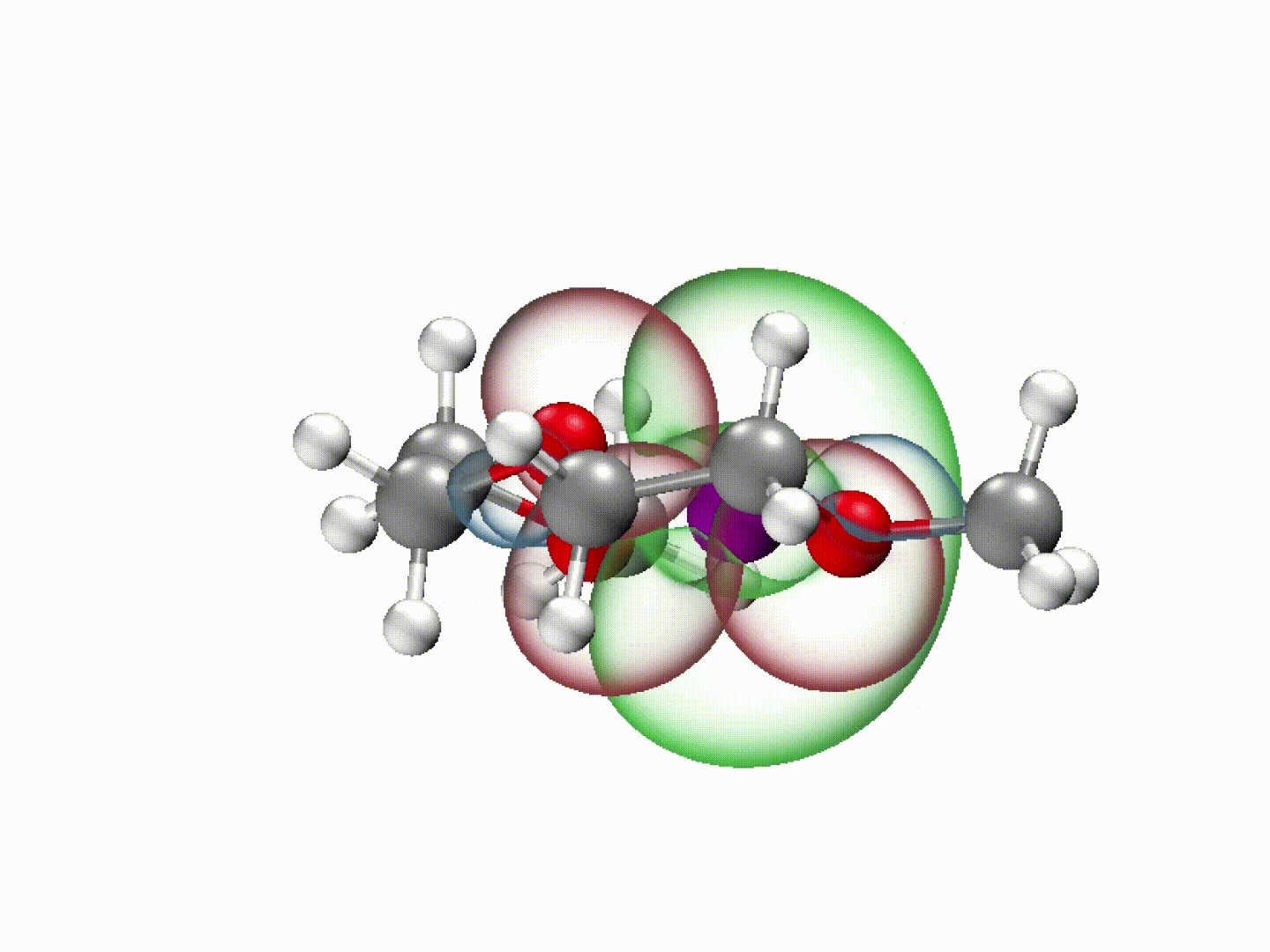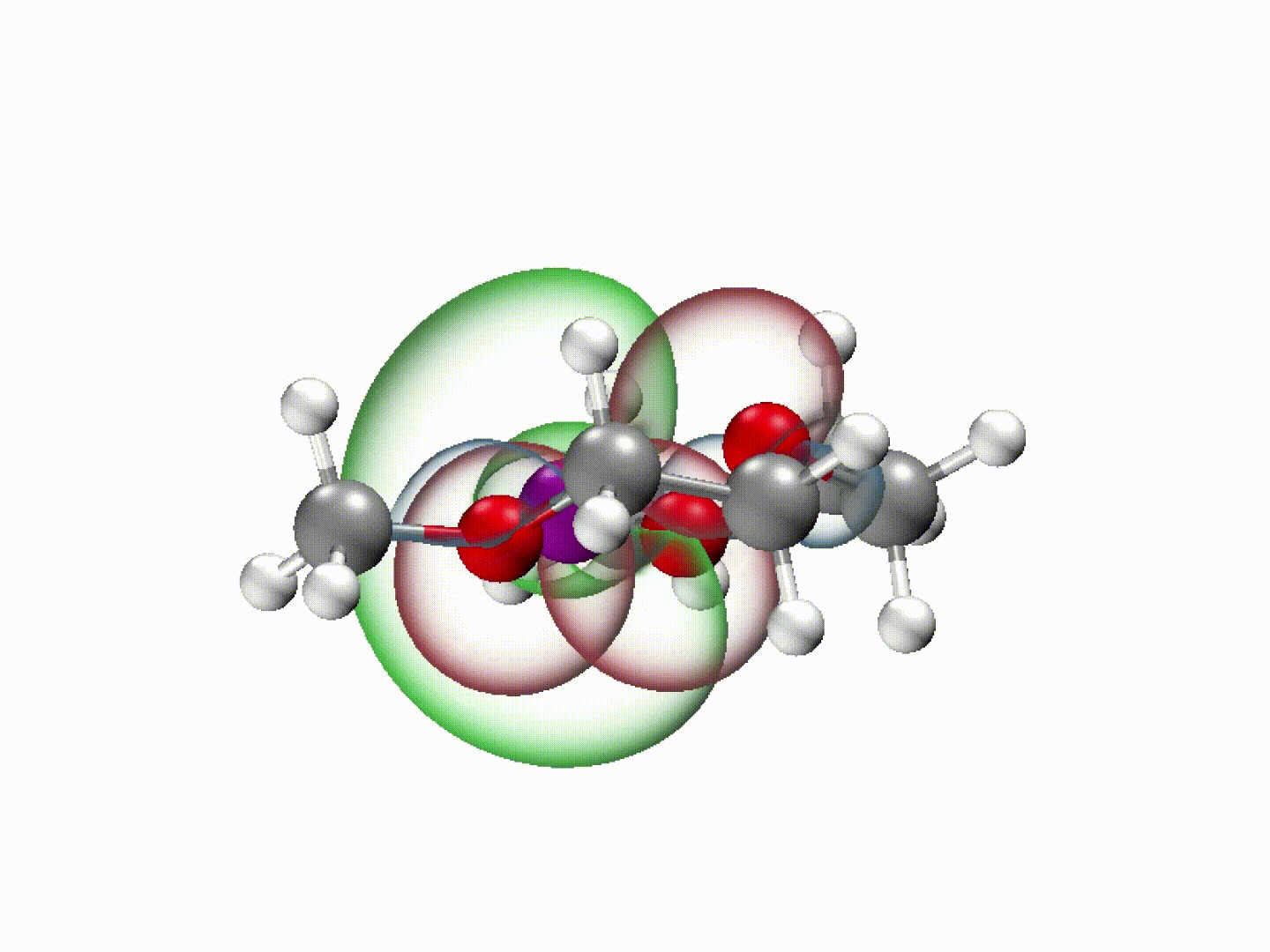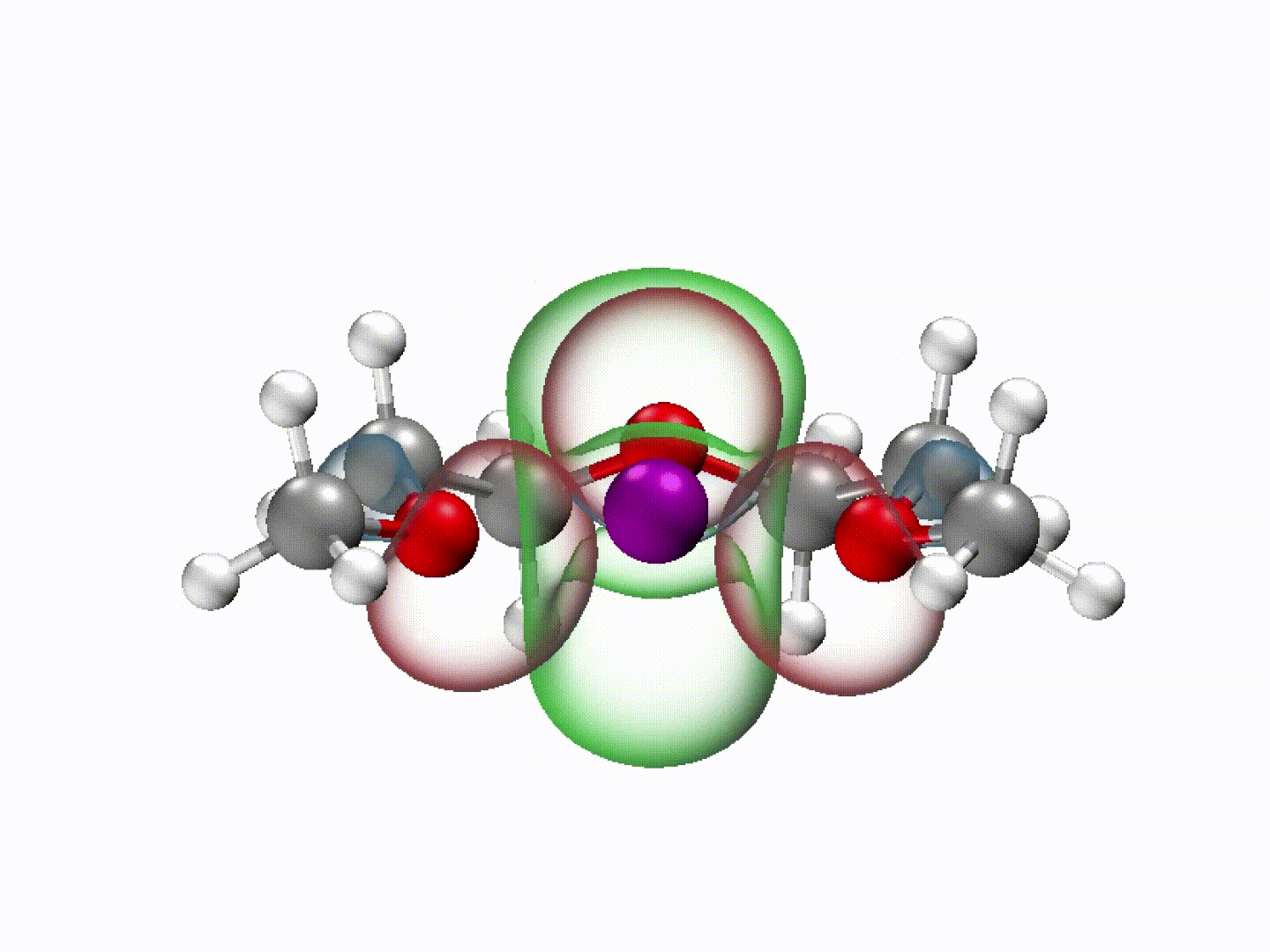
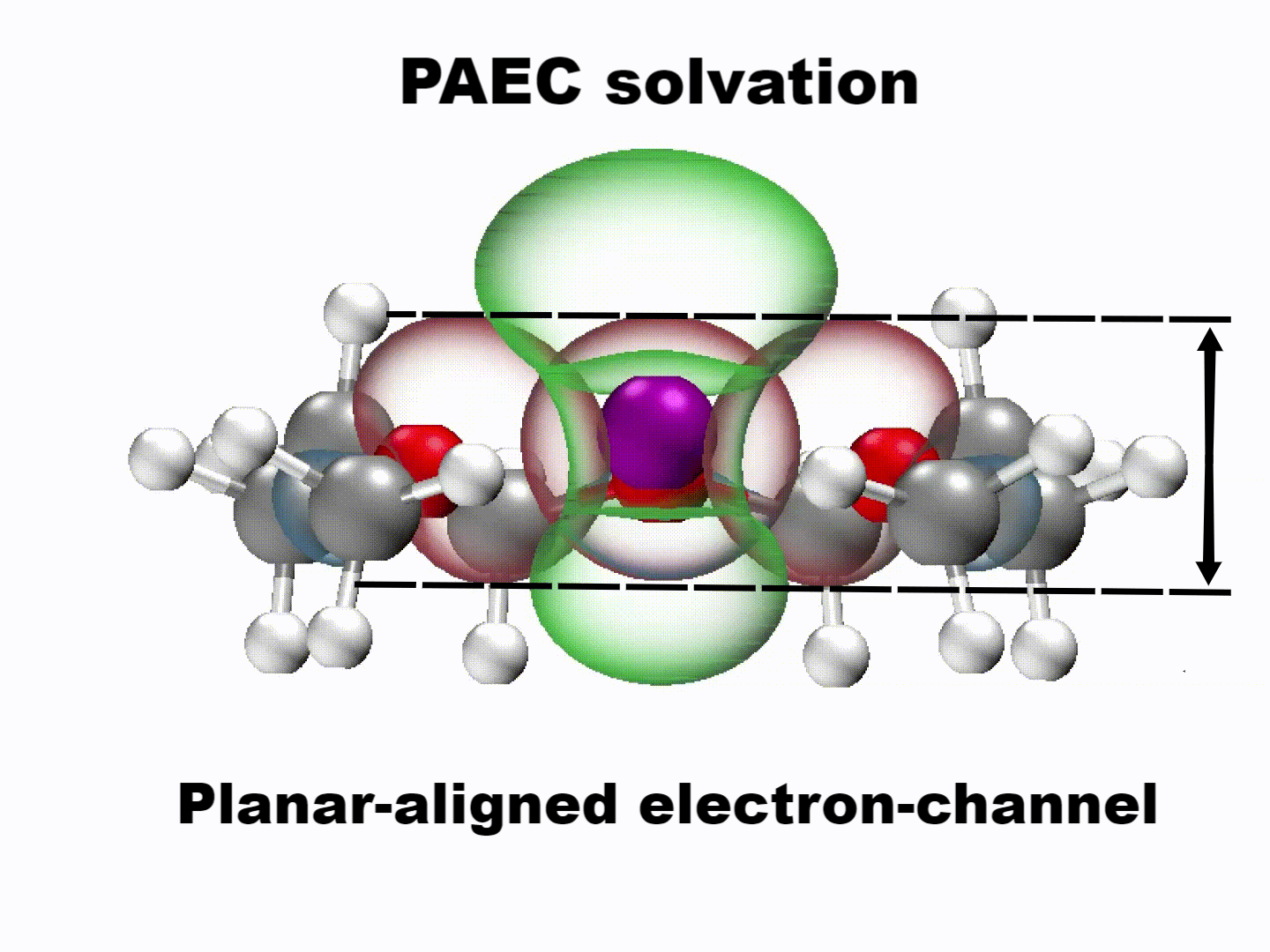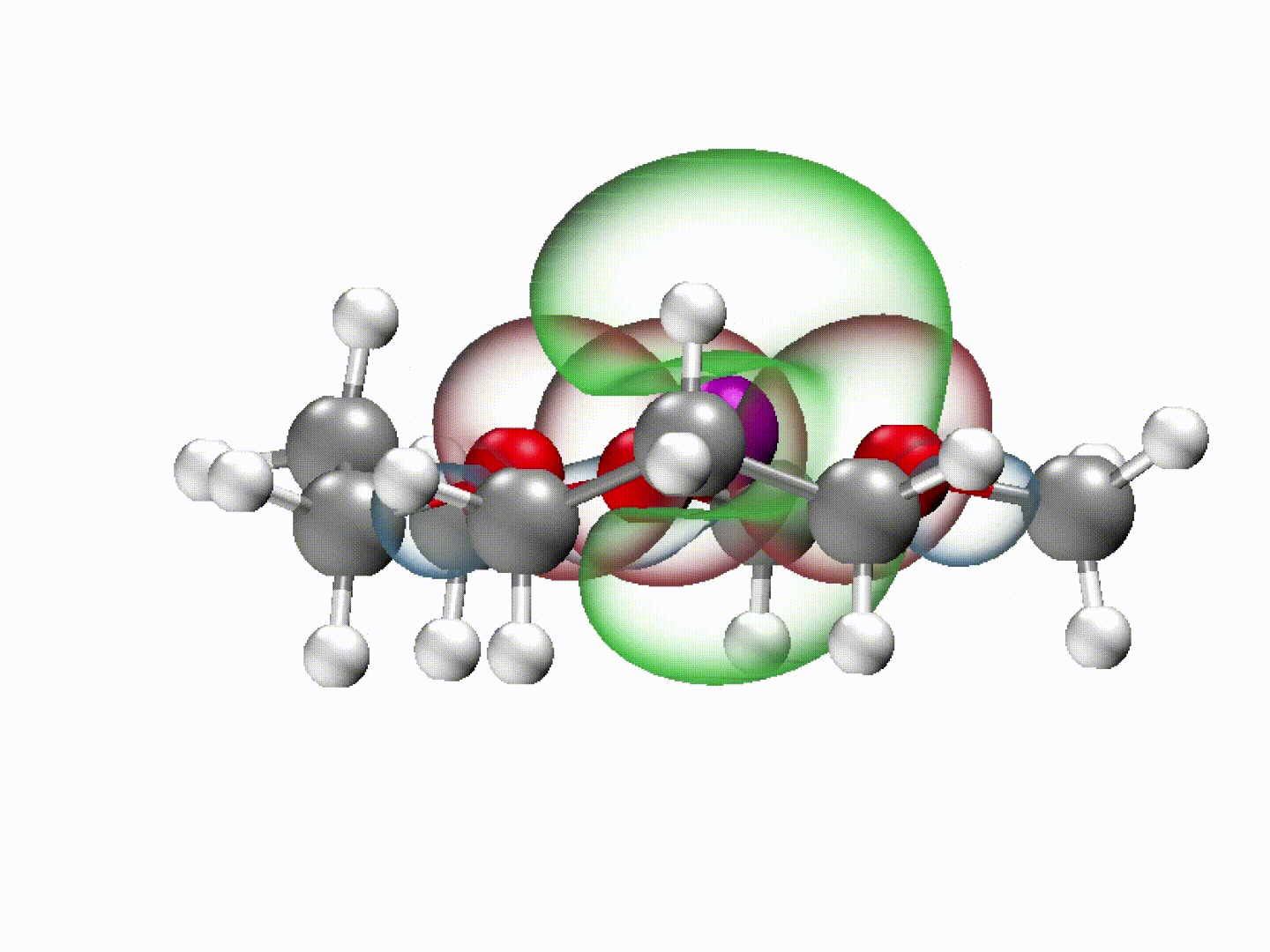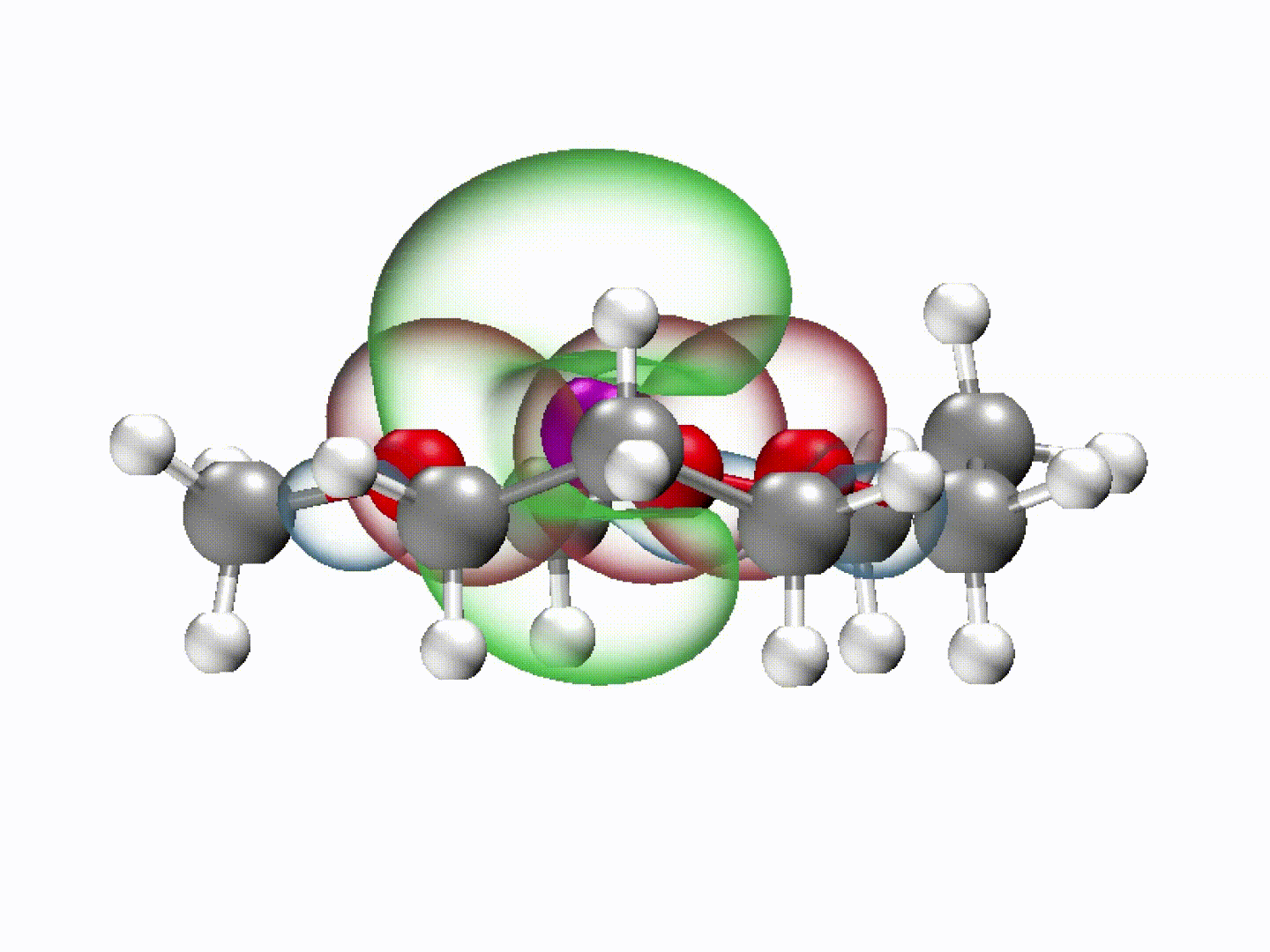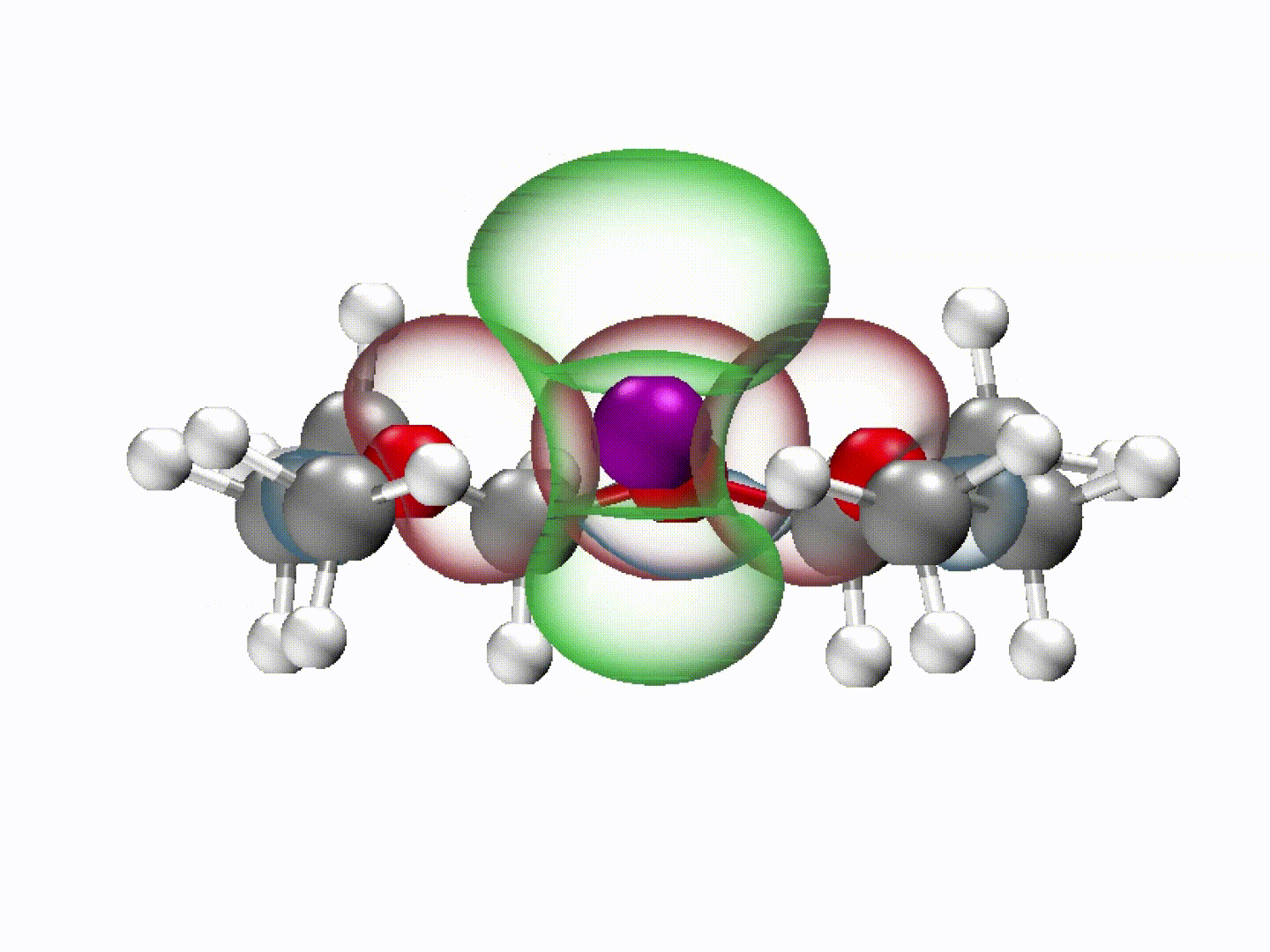
(Dynamic schematic diagram of different solvation structures; red and green regions represent oxygen lone pair electron orbitals and Li+ unoccupied orbitals, respectively.)
One of the most striking aspects of this mechanism is that it challenges a common intuition in electrolyte design based on desolvation model—that weaker solvation makes faster charge transfer. In our case, we found that optimizing the spatial electron distribution outperforms traditional binding regulation, particularly under ultrafast-charging conditions where electron transfer rapidly.
Separating interfacial kinetics from interphase effects
A major challenge in lithium metal research is disentangling intrinsic charge-transfer kinetics from the effects of the solid electrolyte interphase (SEI). Improvements are often attributed to a “better SEI,” even when the underlying mechanism remains unclear.
In this work, we looked at what happens during ultrafast charging, when everything at the battery interface happens in a rush. Under these conditions, the protective SEI layer that usually forms on lithium metal cannot fully develop and is easily broken apart. This leaves the lithium surface largely exposed, allowing the electrolyte itself to directly control fast-charging behavior. In this regime, electrolytes are active players in charge transfer, not just a background medium.
Why this matters
The implications of this finding extend beyond a specific electrolyte formulation or battery chemistry. If molecular alignment in liquid electrolytes can be harnessed to accelerate electron transfer, it opens a new design principle for electrochemical systems.
Rather than relying solely on bulk properties such as viscosity or dielectric constant, future electrolytes could be engineered to self-organize under operating conditions, adapting dynamically to high currents and strong fields. This perspective may be relevant not only for lithium metal batteries, but also for sodium metal batteries, multivalent systems, and other electrochemical technologies where fast interfacial kinetics are critical.
Looking forward
This study represents an early step in exploring how molecular-scale electron organization influences macroscopic battery performance. Many questions remain open. How general is this alignment effect across different solvents and salts? Can we deliberately amplify it through molecular design? And how does it evolve during long-term cycling?
What this work has reinforced for us is the importance of looking at familiar systems from unfamiliar angles. Sometimes, meaningful progress does not come from adding more barriers or stronger interactions, but from allowing molecular electrons to align and move more naturally.
Follow the Topic
-
Nature Energy
Publishing monthly, this journal is dedicated to exploring all aspects of this on-going discussion, from the generation and storage of energy, to its distribution and management, the needs and demands of the different actors, and the impacts that energy technologies and policies have on societies.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in