Molecular guardians of homeostatic synaptic plasticity in Alzheimer’s disease
Published in Neuroscience, Protocols & Methods, and Cell & Molecular Biology
Traumatic brain injury (TBI), including mild or sub-concussive blows, is the largest non-genetic, non-aging-related contributor to Alzheimer’s (AD)-like dementia.1–3 Overall, TBI-cognitive impairments trigger chronic traumatic encephalopathy (CTE), a tauopathy.4,5 Contact sports are a major risk factor for AD.6 Earlier, we observed that concussions in college football players are accompanied by changes in certain serum microRNAs along with behavioral consequences.7
Our studies were guided by the concept that the brain, when confronted with perturbations, such as TBI, rapidly activates tightly regulated resiliency survival programs, triggering the transient formation of protective mediators to counteract the onset of damage. Thus, we explored ischemia-reperfusion injury, as in stroke, and photoreceptor degeneration. These conditions offered windows into neural damage responses. Moreover, we also tested a human disease—age-related macular degeneration (AMD).
We uncovered a docosahexaenoic acid (DHA) metabolite, 10,17S-docosatriene, after 1 hour of middle cerebral artery occlusion in a mouse model8 that reduced leukocyte infiltration, inflammatory gene expression, and infarct size, when isolated and infused into the brain after stroke. We then named this lipid neuroprotectin D1.9-10 Moreover, we found that NPD1 abundance is decreased in the CA1 area in early-stage AD patients.11 A systemic involvement of essential fatty acids in neurodegenerative diseases was suggested by the finding of a blood DHA shortage in Usher’s syndrome patients—a neurosensorial degeneration characterized by blindness and deafness.12
Figure 1 provided the basis for the discovery of ELVs. We identified adiponectin receptor 1 (AdipoR1) as a regulator of photoreceptor cell (PRC) survival through DHA uptake, retention, and conservation.13 We used AdipoR1 knockout (KO) mice made by retroviral gene trapping and homologous recombination and revealed that ablation of this receptor leads to PRC degeneration and absence of 32:6n3 and 34:6 n3 (Fig. 1). So, we explored if the shortage in those VLC-PUFAs would be precursors of novel molecules, which turn out to be ELVs.
These findings prompted a deeper question that shaped this study:14 how does the brain protect itself when its homeostasis is suddenly disrupted?
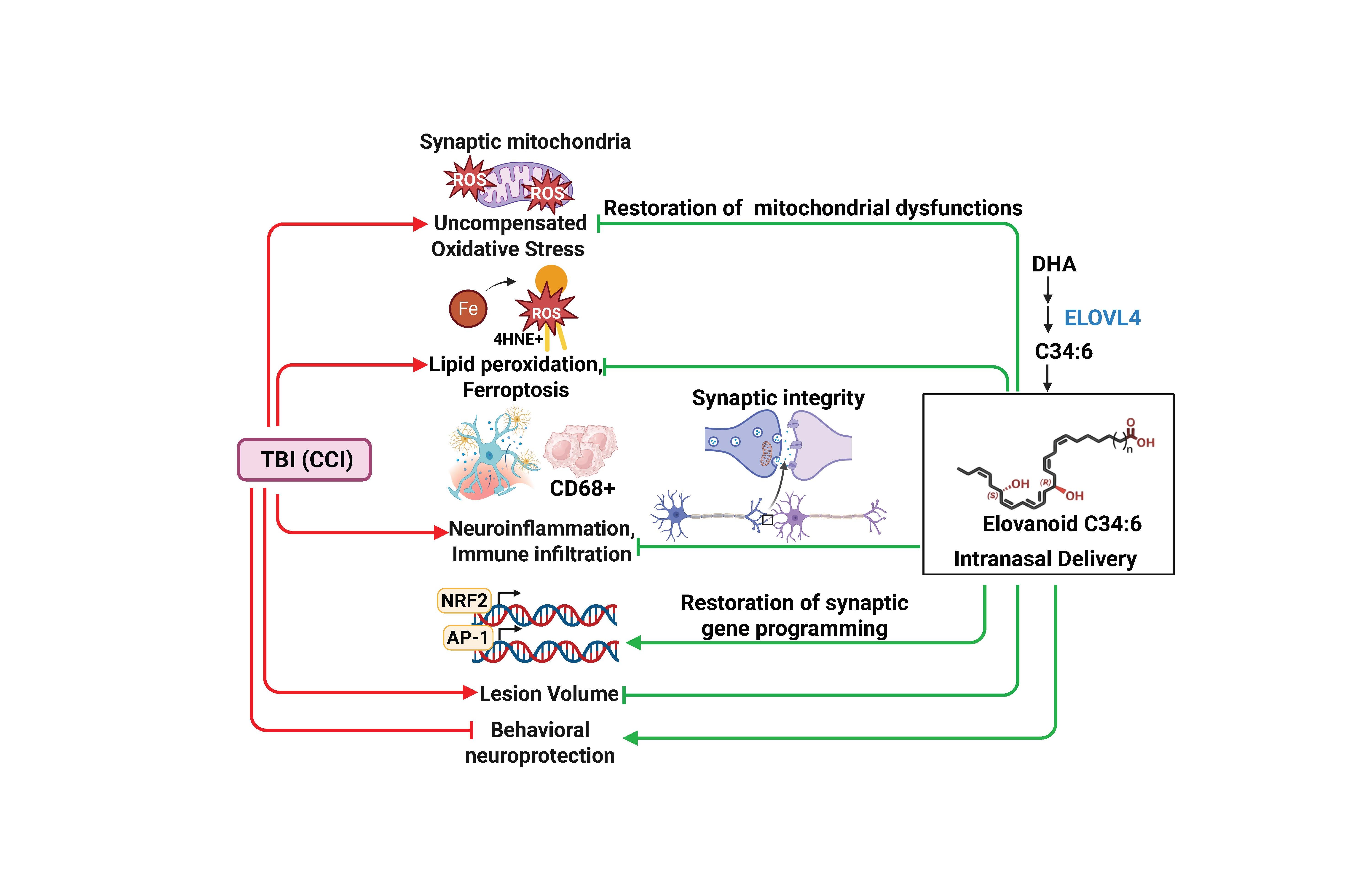
Our current study14 is also based on the demonstration that ELV-mediated protection reduced axonal damage and preserved white matter tracts in two TBI rat models. Furthermore, intranasal ELVs were higher in the ipsilateral cortex compared to the contralateral cortex, showing the effectiveness of targeted delivery.
Unlike early-onset AD, associated with a PSEN1 mutation, most AD cases are late-onset and multifactorial. Cognitive decline and memory loss involve synaptic dysfunctions and AD is a tauopathy.15,16 In response to the onset of damage, the brain defends itself by activating the production of molecular “guardians” to preserve integrity and function.
Elovanoids (ELVs), di-hydroxylated 32:6n3 and 34:6n3, are ELV-N32 and ELV-N34.17,18 ELVs introduce a new aspect of neuroprotection as guardians, serving as an initial line of defense when cell homeostasis is jeopardized.
VLC-PUFAs (C>28) are made by the enzyme ELOVL4 (elongation of very long chain fatty acid-4), selectively expressed in neurons and photoreceptors. Genetic mutations of the gene encoding this enzyme cause loss of vision in Stargardt macular dystrophy type 3, and recessive mutations are associated with intellectual disability, seizures, and spastic quadriplegia. Neural membranes phospholipids contain at the sn-1 position mainly ELV precursors, while DHA, the NPD1 precursor, is at the sn-2 position. Therefore, NPD1 synthesis requires PLA2 to cleave the sn-2 acyl chain to release DHA, whereas ELV synthesis requires PLA1 to cleave the sn-1 acyl chain and release 32:6n-3 and/or 34:6n-3.
The overall bioactivity of ELVs modulates homeostatic synaptic plasticity. It comprises longevity signaling by deacetylase Sirtuin 1 (SIRT1) abundance, as shown in human retinal cells undergoing UOS. ELVs also modulate E3 ubiquitin ligase (Iduna) and the transcriptional regulator prohibitin type-1, which are involved in senescence and cell cycle regulation; stabilize the mitochondrial genome; and modulate mitochondrial biogenesis, the intrinsic apoptotic pathway, and anti-apoptotic proteins, Bcl-2 and Bcl-x. Moreover, ELV synthesis is downregulated in AD models.19

Figure 1. Photoreceptor degeneration leads to the depletion of ELV precursors. (a) AdipoR1 KO mice displayed PRC degeneration and specific retinal DHA reduction and absence of 32:6n-3 and 34:6n-3. (b) Both AdipoR1 and membrane frizzled-related protein (Mfrp) KOs lead to a drastic shortage of 32:6n-3 and 34:6n-3 of retina, making ELV precursors unavailable.
[Source: Bazan 2026 (Wolters Kluwer Health)20 was adapted with permission from Kautzmann et al., 2019 (John Wiley and Sons).21]
The bioactivity of ELVs also includes: a) neuronal protection against O2/glucose deprivation or N-methyl-D-aspartate receptor-mediated excitotoxicity, and b) neuroprotection in experimental ischemic stroke when given 1 hour after 2 hours of ischemia.18,22
Subretinal injection of oligomeric Aβ (OAβ) simulates inflammation in mice retina, resulting in RPE damage and PRC death. OAβ initiates senescence gene programming (p53, p21, p27, p16INK4a, IL-6, senescence-associated secretory phenotype), enhanced expression of autophagy- and AMD-related genes, including human complement factor and extracellular matrix genes, leading to damage of RPE and PRC.23 These events were counteracted by ELVs.
ELVs are autocrine or paracrine mediators that enhance the abundance of cell survival proteins and decrease those engaged in cell death. They also stabilize the mitochondrial genome and modulate mitochondrial biogenesis. Spatial biology has helped in the current study14 to define cell phenotypes, transcriptomics, genomics, and epigenomics that regulate ELV.
ELV-34 modulates uncompensated oxidative stress by regulating TXNRD1 activity, a reductase of TRX, the effector that targets proteins via reduction of –S–S- to –SH–SH (e.g., PTEN). PTEN is altered by ROS, and the oxidative modification it undergoes activates PI3K/AKT signaling, thereby protecting cells from oxidative stress.24 One outcome is that ELV maintains reduced thioredoxin (TXN) using NADP) H + H+ as a proton donor and FAD as an electron acceptor to sustain homeostasis.25
AMD and AD accumulate Aβ in the retina and brain, respectively. OAβ triggers RPE and PRC pathology.23 Rods in the retina periphery of AMD females displayed a shortage of ELV precursors. Estrogen receptor β is expressed in the retina and modulates inflammatory responses. About 66% of people with AMD are female.26 Therefore, women become susceptible to retinal degeneration with age and gradual estrogen reduction, hence limiting the generation of protective ELVs.
Dietary omega-3 fatty acids or their supplements need to overcome physiological hurdles before arriving in the brain. Firstly, the role of the gastrointestinal tract in the handling of omega-3 fatty acids is complex due to the changing microbiome during different ages. Then omega-3 fatty acids must go through the liver, where they become part of lipoproteins.27 Then, receptors take up the omega-3 fatty acids from the bloodstream in the nervous system and further metabolism occurs, such as enzyme-mediated elongation and desaturation.
Single-cell multiome and spatial transcriptomics reveal synaptic deconstruction underlying early AD tauopathy.28 The current study14 used single-cell RNA sequencing and spatial transcriptomics to map brain cell-cell communication networks by CellChat and found altered brain-derived neurotrophic factor networks and increased Netrin signaling. NeuronChat predicted glutamatergic disruptions (https://go.nature.com/478drqj). The integration of transcriptomics and epi-transcriptomics provides a framework for the complex cellular dynamics in brain damage responses.
In conclusion, ELVs are molecular guardians of the nervous system, functioning as low-abundance, high-potency mediators made on demand that modulate homeostatic synaptic plasticity. Diets enriched in DHA reversed cognitive decline or sight loss, suggesting that AD and AMD involve a DHA shortage. Therefore, diet plays a critical role in healthy aging by providing ELVs precursors that, upon conversion into ELVs, exert protection spanning stroke, TBI, macular degeneration, Alzheimer’s, and Parkinson’s. As an early line of defense, ELVs initiate pro-homeostatic and protective resiliency through distinct targets, including sustaining antioxidant capacity by modulation of thioredoxin reductase 1 and the glutathione system, positioning them as regulators of neural longevity and disease resistance. Translational advances of ELV bioactivity would contribute to open avenues for therapeutic development.
References
- Graham, A., Livingston, G., Purnell, L. & Huntley, J. Mild Traumatic Brain Injuries and Future Risk of Developing Alzheimer’s Disease: Systematic Review and Meta-Analysis. J Alzheimers Dis 87, 969–979 (2022).
- Plassman, B. L., Chanti-Ketterl, M., Pieper, C. F. & Yaffe, K. Traumatic brain injury and dementia risk in male veteran older twins-Controlling for genetic and early life non-genetic factors. Alzheimers Dement 18, 2234–2242 (2022).
- Padron, D. & Carballea, D. Systematic Review on the Association of a Traumatic Brain Injury (TBI) and Development of Alzheimer’s Disease (AD). Archives of Physical Medicine and Rehabilitation 103, e5–e6 (2022).
- Arena, J. D. et al. Traumatic brain injury or head impacts from contact sports are associated with tau astrogliopathy. Brain 148, 2671–2683 (2025).
- Kumar, M. et al. Molecular features of human pathological tau distinguish tauopathy-associated dementias. Cell 189, 956-968.e13 (2026).
- Butler, M. L. M. D. et al. Repeated head trauma causes neuron loss and inflammation in young athletes. Nature 647, 228–237 (2025).
- Wyczechowska, D. et al. Serum microRNAs associated with concussion in football players. Front Neurol 14, 1155479 (2023).
- Marcheselli, V. L. et al. Novel Docosanoids Inhibit Brain Ischemia-Reperfusion-mediated Leukocyte Infiltration and Pro-inflammatory Gene Expression. J. Biol. Chem. 278, 43807–43817 (2003).
- Mukherjee, P. K., Marcheselli, V. L., Serhan, C. N. & Bazan, N. G. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 101, 8491–8496 (2004).
- Mukherjee, P. K. et al. Neurotrophins enhance retinal pigment epithelial cell survival through neuroprotectin D1 signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 13152–13157 (2007).
- Lukiw, W. J. et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest 115, 2774–2783 (2005).
- Bazan, N. G., Scott, B. L., Reddy, T. S. & Pelias, M. Z. Decreased content of docosahexaenoate and arachidonate in plasma phospholipids in Usher’s syndrome. Biochem Biophys Res Commun 141, 600–604 (1986).
- Rice, D. S. et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat Commun 6, 6228 (2015).
- Giles, B. L. et al. Elovanoid neuroprotection targets cell transcriptomics and proteomics to sustain synaptic integrity after brain injury. Commun Biol https://doi.org/10.1038/s42003-026-09931-1 (2026) doi:10.1038/s42003-026-09931-1.
- Dubois, B. et al. Alzheimer Disease as a Clinical-Biological Construct-An International Working Group Recommendation. JAMA Neurol https://doi.org/10.1001/jamaneurol.2024.3770 (2024) doi:10.1001/jamaneurol.2024.3770.
- Mathys, H. et al. Single-cell multiregion dissection of Alzheimer’s disease. Nature 632, 858–868 (2024).
- Jun, B. et al. Elovanoids are novel cell-specific lipid mediators necessary for neuroprotective signaling for photoreceptor cell integrity. Sci Rep 7, 5279 (2017).
- Bhattacharjee, S. et al. Elovanoids are a novel class of homeostatic lipid mediators that protect neural cell integrity upon injury. Sci. Adv. 3, e1700735 (2017).
- Swinkels, D. et al. DHA Shortage Causes the Early Degeneration of Photoreceptors and RPE in Mice With Peroxisomal β-Oxidation Deficiency. Investigative Ophthalmology & Visual Science 64, 10–10 (2023).
- Bazan, N. G. Elovanoids: linking nutrition to neuroprotection. Curr Opin Clin Nutr Metab Care 29, 111–122 (2026).
- Kautzmann, M.-A. I. et al. Membrane-type frizzled-related protein regulates lipidome and transcription for photoreceptor function. The FASEB Journal 34, 912–929 (2020).
- Belayev, L. et al. Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ 24, 1091–1099 (2017).
- Do, K. V. et al. Elovanoids counteract oligomeric β-amyloid-induced gene expression and protect photoreceptors. Proc Natl Acad Sci USA 116, 24317–24325 (2019).
- Zhang, Y. et al. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol 34, 101553 (2020).
- Calandria, J. M. et al. Elovanoid-N34 modulates TXNRD1 key in protection against oxidative stress-related diseases. Cell Death Dis 14, 819 (2023).
- U.S. National Institutes of Health, N. E. I. Eye Health Data and Statistics. National Eye Institute https://www.nei.nih.gov/learn-about-eye-health/eye-health-data-and-statistics (2022).
- Scott, B. L. & Bazan, N. G. Membrane docosahexaenoate is supplied to the developing brain and retina by the liver. PNAS 86, 2903–2907 (1989).
- Do Carmo, S. et al. Differential effect of an evolving amyloid and tau pathology on brain phospholipids and bioactive lipid mediators in rat models of Alzheimer-like pathology. J Neuroinflammation 21, 185 (2024).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Congrats! This is a very novel approach linking several neuropathologies.