MYT1L haploinsufficiency in human neurons and mice causes autism-associated phenotypes that can be reversed by genetic and pharmacologic intervention
Published in Neuroscience
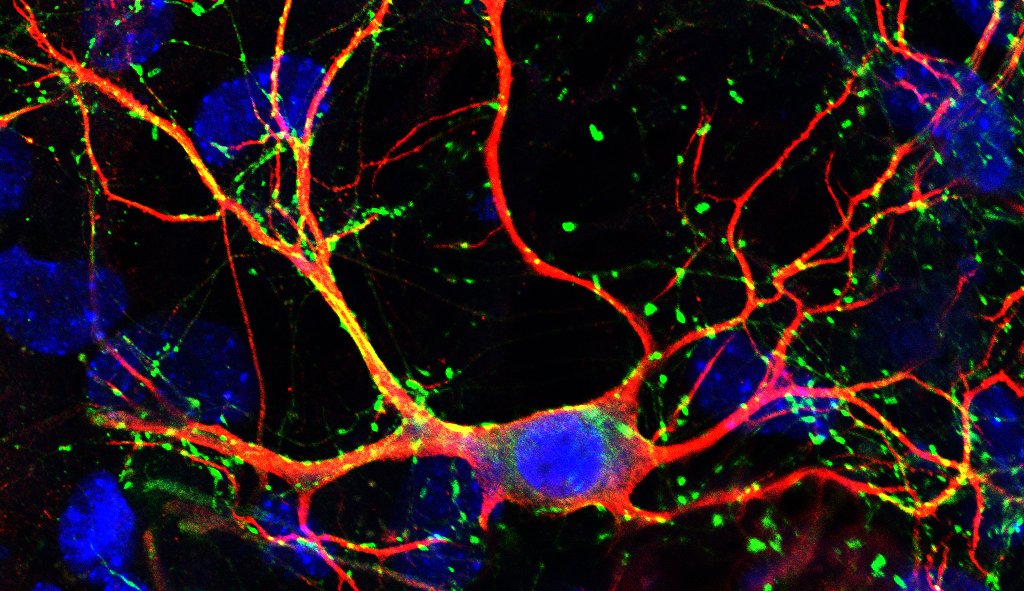

The development of the human brain is a very complex process, which relies on a closely-controlled interplay between several signalling molecules and transcription factors. This complexity renders it sensitive to disruption, which can cause neurodevelopmental defects. Neurodevelopmental disorders (NDDs), which include autism spectrum disorder (ASD), intellectual disability (ID), attention deficit hyperactivity disorder (ADHD), epilepsy, and schizophrenia, constitute a huge global health burden. However, the pathogenesis of these disorders remains poorly understood, making it very difficult to find targeted treatment options. Recent studies revealed that transcriptional regulators can contribute to the development of NDDs. Among these, the zinc finger transcription factor MYT1L stands out as as being expressed in virtually all neurons throughout life (Cardoso-Moreira et al., 2019, GTEx Portal, Mansfield et al., 2020).
MYT1L was originally identified as a strong reprogramming factor that, in combination with two pro-neural transcription factors is capable of directly reprogramming fibroblasts into functional neurons (Vierbuchen et al., 2010). Subsequent studies showed that this neuron-specific factor promoted neuronal identity by repressing negative regulators of neurogenesis, as well as transcriptional regulators of several non-neuronal lineages (Mall et al., 2017). MYT1L mutations are strongly associated with several NDDs, with 98% of currently reported cases with heterozygous MYT1L loss of function mutations being diagnosed with ASD and/or ID (Mansfield et al., 2020). Recently-published MYT1L mouse models (Chen et al., 2021, Kim et al., 2022, Wöhr et al., 2022) showed that MYT1L mutations can cause NDD-associated phenotypes in mice. However, the mechanisms by which MYT1L mutations contribute to disease phenotypes remain elusive. Furthermore, no study presents the molecular effects of MYT1L depletion in human neurons or any potential therapeutic interventions for MYT1L syndrome patients.
Our study used genetically-engineered mice and human induced neurons to investigate how MYT1L regulates brain development, whether mutations are sufficient to cause NDDs, and if resulting phenotypes can be targeted therapeutically. Our findings have recently been published in Molecular Psychiatry under the title “MYT1L haploinsufficiency in human neurons and mice causes autism-associated phenotypes that can be reversed by genetic and pharmacologic intervention”.
We found that MYT1L deficiency caused upregulation of bound target genes, including negative regulators of neurogenesis, like members of the WNT and NOTCH signalling pathways. Consequently, we observed neurodevelopmental delays that could be partially rescued by chemical inhibition of these pathways. MYT1L-deficient mice presented with abnormal brain morphology and behavioural deficits. Homozygous mice died after birth, emphasising that MYT1L is crucial for normal brain development and survival. In addition to developmental defects, we detected a striking network hyperactivity and a strong upregulation of non-neuronal genes in mouse and human neurons upon MYT1L depletion. This was very exciting to see because it indicates that MYT1L is needed to switch off unwanted genes in neurons, not only during direct reprogramming but also during normal differentiation. Losing this safeguarding function of MYT1L seemed to result in the development of neurons with a "confused" transcriptional identity. In the search for a connection between this transcriptional confusion and functional deficits, we found the main cardiac sodium channel SCN5A to be significantly upregulated upon MYT1L depletion in mouse and human neurons. Upregulation of this channel, which should normally not be expressed in high levels in the brain, offers a possible explanation for the unexpected neuronal network hyperactivity. Supporting this hypothesis, we were able to normalise electrophysiological hyperactivity in vitro by Myt1l overexpression and Scn5a knockdown. Excitingly, the FDA-approved sodium channel blocker lamotrigine rescued not only electrophysiological abnormalities in vitro but also behavioural deficits in vivo. This suggests that functional hyperactivity of neurons can, at least in part, provoke behavioural abnormalities in mice, which can be treated after neurodevelopment is complete.
All in all, we present evidence that MYT1L depletion is sufficient to cause NDD-associated phenotypes in mouse and human models. While similar observations have previously been described for other MYT1L mouse models, our study is the first to confirm these findings in human neurons. We show that MYT1L is crucial for the repression of non-neuronal genes in neurons, which might represent a novel mechanism that, at least in part, contributes to the aetiology of NDDs. Transcriptional deregulation results in neurodevelopmental defects and post-natal phenotypes, including electrophysiological hyperactivity of neurons and behaviour deficits in mice. These phenotypes can partially be rescued in post-mitotic neurons and adult mice, which emphasises the important role of MYT1L not only during development but also after neurogenesis. This makes MYT1L stand out compared to many other NDD-associated gene regulators, which often primarily act during neurodevelopment and opens the possibility that therapeutic intervention can benefit patients with MYT1L syndrome at the time of diagnosis, which is usually after neurodevelopment is complete.
One limitation of this study is that we only investigated the effect of MYT1L loss. For MYT1L syndrome patients, not only loss-of-function variants but also missense variants and duplications of MYT1L have been reported. The broad range of patient phenotypes indicates that these mutations disrupt neuronal function differently. With modern technologies, we can engineer stem cells to carry patient-specific mutations or generate patient-derived neurons from induced pluripotent stem cells, which allows us to study how specific mutations or genetic backgrounds affect disease aetiology.
The discovery of MYT1L as a life-long safeguard of neuronal identity and its crucial role in normal brain function raises new exciting questions. What happens if MYT1L is removed after neurodevelopment in postmitotic neurons? Interestingly, reduced MYT1L expression and loss of neuronal cell identity have been suggested to play a role in Alzheimer’s disease models (Caldwell et al., 2020, Mertens et al., 2021). Given that MYT1L expression decreases during aging, it is tempting to speculate that life-long repression of non-neuronal genes in neurons could also play a role in the aetiology of neurodegenerative diseases. Furthermore, transcription factors with similar safeguarding functions might exist in other cell types, and the concept of silencing unwanted cell fate signatures might be a general mechanism disrupted in disease.

This work was supported by funding from the Chica and Heinz Schaller Stiftung, 2019 NARSAD Young Investigator Grant, DFG SFB1158-SO2, a 2021 Fritz Thyssen Grant, CellNetworks (EXC81), 2020 NARSAD Young Investigator Grant, DFG 504019642, ERC StG No 804710, DFG SFB1324 and the Hector Stiftung II gGmbH. Fellowships were provided by the DAAD/ ANID (57451854/62180003) and the Helmholtz International Graduate School. Open Access funding enabled and organized by Projekt DEAL. The illustration was made using Biorender.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.

Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcement

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in